
24일 레켐비, 경도인지장애 및 초기 알츠하이머병 대상 허가
MSD '툴리소키바트', 활성 크론병 환자 대상 3상 임상시험계획 승인
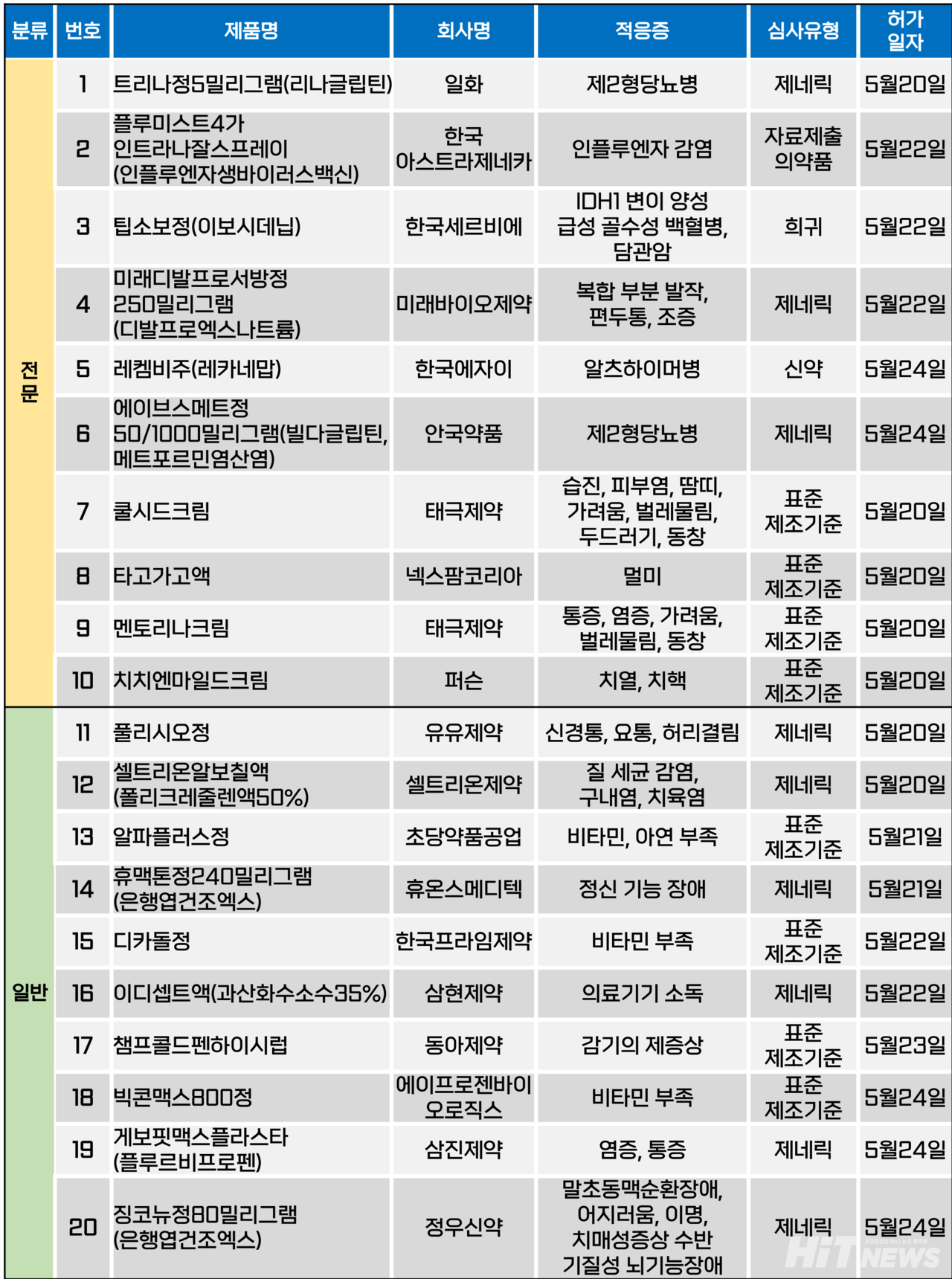
지난주(5월 20일~24일) 총 20개 품목이 식품의약품안전처로부터 품목허가를 받았다. 전문의약품 및 일반의약품 모두 각 10개 품목 씩이다. 이들은 제2형당뇨병, 인플루엔자 감염, 급성 골수성 백혈병, 복합 부분 발작, 알츠하이머병 등 다양한 적응증으로 허가됐다.
에자이가 개발한 레켐비주(성분 레카네맙)가 알츠하이머병으로 인한 경도인지장애 및 초기 알츠하이머병을 대상으로 지난 24일 허가됐다. 미국, 일본, 중국 등에 이어 4번째 허가다.
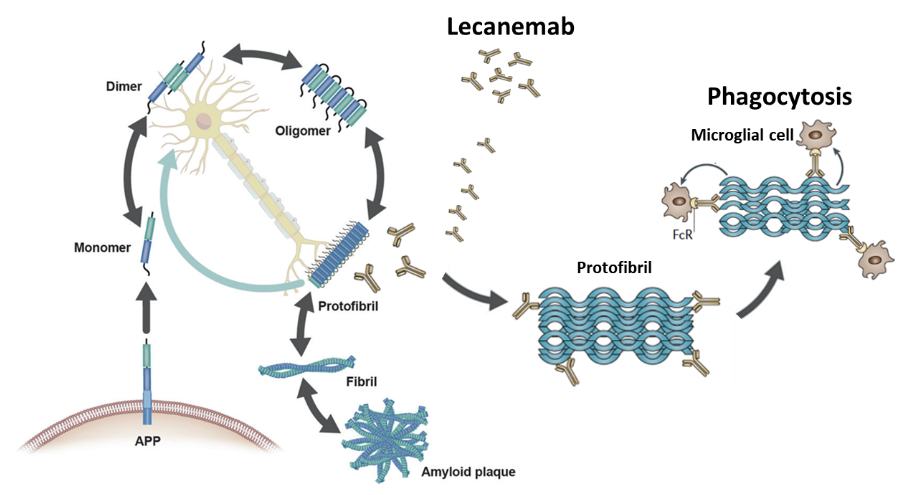
레켐비는 뇌 내 아밀로이드 베타(Aβ) 응집체(aggregates)와 원섬유(protofibrils)에 선택적으로 결합해 이들을 제거하는 인간화 면역글로불린 G1(IgG1) 단일클론항체 의약품으로, 인지기능 소실 등 질병 진행을 늦추는 작용기전을 가진다. 그동안 국내 허가된 알츠하이머병 치료제는 대증요법에 의한 증상 완호를 목적으로 한 제품이 뿐이었다.
이번 허가의 근거가된 주요 3상 임상은 'Clarity AD' 연구다. 연구 결과, 레켐비는 18개월 시점에 위약군 대비 CDR-SB(Clinical Dementia Rating Sum of Boxes)을 0.45점 감소시켜, 알츠하이머병 진행을 27% 지연 시키는 것으로 나타났다(95% CI : -0.67~-0.23, P<0.001). 다만, 초기 알츠하이머병 외 중등도 이상 환자에 대한 효과와 안전성은 아직 확인되지 않았다.
레켐비는 '아밀로이드-관련 영상 이상(Amyloid -Related Imaging Abnormality· ARIA)'이 발생할 수 있는 것으로 나타났는데, ARIA는 영상 검사에서 확인되는 뇌부종 또는 미세출혈 등의 비정상적인 방사선 소견을 말한다.
식약처 관계자는 "ARIA는 대체로 무증상이나, 두통, 혼돈, 어지러움 등이 발생할 수 있으며 대개 시간이 지남에 따라 소실된다"면서 "다만, 드물게 발작 및 뇌전증 등 중대하고 생명을 위협하는 사례가 발생할 수 있어 허가 사항에 따른 자기공명영상 모니터링(MRI) 등의 정기적 영상 모니터링이 필요하다"고 설명했다.
한편, 에자이는 바이오젠과 레켐비 상업화 및 프로모션에 대한 협력을 체결한 바 있으며, 전 세계 레켐비 개발 및 규제 제출, 상업화 및 프로모션에 대한 최종 의사결정 권한을 갖고 있다. 국내에서는 한국에자이가 레켐비 제품 유통 및 홍보·마케팅을 주도할 예정이다.
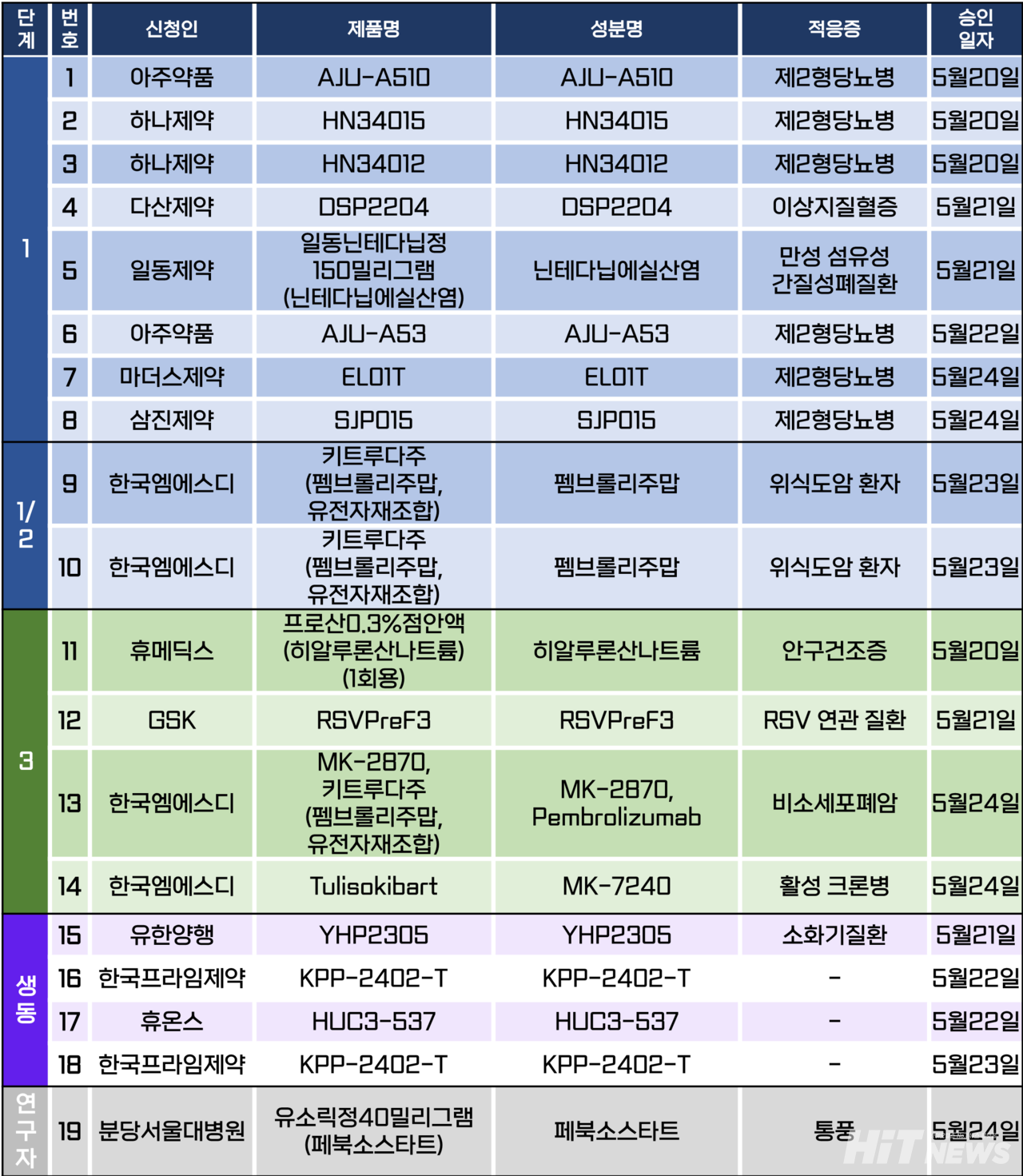
임상시험계획은 총 19건이 승인됐다. 세부적으로 △1상 8건 △1/2상 2건 △3상 4건 △생물학적 동등성 4건 △연구자임상 1건 등이다. 이 임상시험들은 제2형당뇨병, 이상지질혈증, 만성 섬유성 간질성폐질환, 안구건조증, 활성 크론병 등 질환과 위식도암, 위장관암, 췌장관선암종, 소세포폐암 등 암종을 대상으로 승인됐다.
미국 머크(MSD)가 개발 중인 활성 궤양성 대장염 및 크론병 치료 신약 후보물질인 '툴리소키바트(Tulisokibart, 연구코드명 MK-7240)'의 3상 임상시험계획이 지난 24일 승인됐다.
이번 임상 3상은 ' 중등증 내지 중증의 활성 크론병 ' 환자를 대상으로 툴리소키바트의 유효성과 안전성을 평가하기 위한 무작위 배정, 이중 눈가림, 위약 대조 프로그램 연구다.
MK-7240은 종양괴사인자인 'TNK 유사 리간드 1A(TL1A)'을 타깃하는 인간화 단일클론항체로, 궤양성 대장염과 크론병 등 염증성 장질환을 타깃하는 물질로 알려져 있다.
이번 임상은 44명의 중등증 내지 중증의 활성 크론병 환자(글로벌 1200명)을 목표로 오는 2033년 2월까지 △연세대 의대 세브란스병원 △삼성서울병원 △강북삼성병원 △연세대 원주세브란스기독병원 △영남대병원 △서울대병원 △가톨릭대 성빈센트병원 △서울아산병원 △중앙대병원 △인제대 해운대백병원 △동아대병원 △가톨릭대 대전성모병원 △가톨릭대 은평성모병원 △경희대병원 등 의료기관에서 진행될 예정이다.
연구진은 일차유효성평가변수(Primary endpoint)를 미국 FDA, 유럽 EMA 등 규제기관별로 다르게 설정했다. FDA의 경우에는 '12주 및 52주 시점에서의 크론병 활동도 지수 점수에 따른 임상적 관해', EMA의 경우에는 '12주 및 52주 시점에서의 배변 빈도와 복통 통증 점수에 따른 임상적 관해'로 설정했다. 이상사례 및 이로 인한 시험 중재의 중단은 공통 일차유효성평가변수였다.
한편, MK-7240은 MSD가 지난 4월 바이오 기업 프로메테우스를 인수하며 획득한 파이프라인이다. 인수 규모는 108억달러였다. 이 물질은 프로메테우스가 'PRA023'라는 연구명으로 개발하고 있었는데, MSD가 프로메테우스 인수 후 명칭을 MK-7240으로 바꿔 연구를 지속하고 있다.