
히터뷰 |
최환호 퀀텀인텔리전스 대표
생물학을 공부하는 학부생들은 화학과 물리를 함께 배운다. 생물을 설명하려면 화학이, 화학을 설명하려면 물리가 필요하기 때문이다. 따라서 물리법칙을 알면 생물 내에서 벌어지는 현상은 모두 설명 가능하고 예측까지 가능할지 모른다. 만약 모든 원자의 위치와 운동량을 알고 있는 엄청난 존재가 있다면 말이다. 철학자들은 이 가상의 존재를 '라플라스의 악마(Laplace's Demon)'라 부른다.
물론 양자역학의 기본적인 법칙에 따르면 이런 존재는 있을 수 없다. 하지만 알고 싶은 부분을 어느 정도 타협하면, 예컨대 '타깃 X에 붙는 신약 후보물질의 구조를 예측하고 싶다'면, 현대 과학자들이 만들어낸 소환의식을 통해 라플라스의 악마가 반쯤은 강림하게 할 수도 있다. 그 소환 의식이 바로 양자컴퓨팅이다.
멀리 찾아볼 것도 없이, 양자컴퓨팅을 통한 신약개발은 여기 한국에서도 발화했다. 물리-화학-생물을 넘나드는 차세대 신약개발 기술은 이미 코앞에 있다. 양자컴퓨팅 기반 후보물질 발굴 플랫폼을 개발 중인 회사, 퀀텀인텔리전스의 최환호 대표를 만나 이야기를 나눴다.

명함의 M.D., Ph.D. 말이죠. 의사 자격증에 박사 학위를 들고 산업계로 오는 케이스는 드뭅니다. 심지어 양자컴퓨팅이 퀀텀인텔리전스의 주력 기술이군요. 계기가 무엇일까요?
"원래 서울대 물리학과를 졸업했고, 그 다음 의대를 간 뒤 옥스포드에서 화학으로 박사를 했어요. 창업 전에 이렇게 공부했던 데에는 대단한 목표가 있진 않았죠. 솔직히 그냥 흘러가는 대로 살았다 할까요(웃음)."
흘러가는 대로 사시다가 서울대, 의대, 옥스포드까지…다른 분들이 들으면 신기해할 것 같습니다(웃음). 공부한 학문이 계속 바뀌었다는 것도 흥미롭네요.
"물리학이 좋아서 물리학과로 갔다가, 의술이 궁금해져서 의대도 가고, 그랬던 거죠. 이쪽으로 가서 공부했을 때 저쪽을 바라보면 어떨까, 저쪽으로 가서 이쪽으로 보면 어떨까, 하는 생각이었고요.
물리는 워낙 근본적인 원리를 다루는 것이다 보니 응용성이 더 강한 학문을 하고 싶더라구요. 생체 분자 간 인터렉션(상호작용)을 재미있게 공부하다 보니, 화학 구조에 대한 이해가 필요하겠다는 생각이 들었어요. 그래서 결국 화학 박사까지 가게 됐어요."
신약개발 비즈니스로 들어가려고 공부했던 면모도 있었나요?
"그렇진 않았어요. 창업 계기는 후반에 생겼거든요. 신약 관련한 연구도 박사과정 중에 했었고, 그게 아스트라제네카(Astrazeneca)와 진행했던 항생제 개발이었어요. 박테리아가 항생제를 분해해서 내성을 발달하지 못하도록, 항생제 화학구조를 안정화시키는 연구였습니다.
그러니까 쭉 돌아보면 이론에서 실용으로 점점 넘어가는 식으로 공부했던 거죠. 저와 공동창업한 강홍석 CTO(최고기술책임자)만 해도 서울대 물리학과를 나와서 메릴랜드 대학에서 생물리학 박사를 했어요. 저랑 논문을 15편 정도 썼는데, 이 논문이란 것이 논문으로 끝나는 경우가 많아요. 어느 지점에선 논문이 자기만족을 위한 것이 아닌가, 하는 생각이 들더군요."
실용에 대한 갈증이 신약개발로 이어진 데에는, 시장에서 어떤 미충족 수요를 봤기 때문이겠죠.
"학계에 있던 시기에 '오토닥(AutoDock)' 같은 도킹 프로그램(Docking programㆍ분자 간 결합을 예측하는 프로그램)이 쏟아져 나왔어요. 머크(Merck)같은 제약사들도 그런 프로그램을 자체적으로 갖고 있죠. 그런데 이런 프로그램들이 잘 안 맞아요."
무엇이 잘 안 맞죠?
"예를 들어 항체가 항원에 어떤 식으로 붙는지, 단백질 타깃에 저분자 화합물이나 펩타이드가 잘 붙는지 제대로 예측하지 못한다는 거죠. Kd값(해리상수ㆍ분자 간 결합력을 나타내는 지표 중 하나) 같은 걸 예측하면 대부분 빗나갑니다.
그럴 수밖에 없는 게, 단백질이나 펩타이드 같은 복잡한 분자들은 레고 블록처럼 가만히 모양을 유지하지 않아요. 꿈틀꿈틀, 흐느적거리면서 돌아다니거든요. 이렇게 자유도가 높은 구조의 행동을 예측한다는 건 일반적인 컴퓨터로는 잘 안 돼요. 종국엔 양자컴퓨터가 필요해집니다."
즉 분자의 행동을 예측하려면 너무나 많은 경우의 수를 계산하는 능력이 필요한데, 그런 연산능을 갖추려면 양자컴퓨터로 가야 한다는 이야기.
"그렇죠. 기본적으로 생물학이란 게 이미 만들어져 있는 걸 설명하는 학문이라서 그래요. 예측하려면 복잡한 생체를 어느 정도 단순화해서 설명해야 해요. 문제는 그 단순화가 지나칠 수 있다는 거죠. 그러다 보니 뭔가 예측한다는 게 힘듭니다.
반대로 물리학을 보면요. 이미 만들어져 있는 걸 설명한다기보단, 기초적인 이론에서 시작해서 만들어 나가는 학문에 가까워요. 원리, 이론, 수식이 맞다면 이것으로 예측된 현상은 다 맞는 거예요. 뉴턴 할아버지가 와도요(웃음).
그래서 생물학과 물리학 사이에는 벽이 있어요. 생물학도들은 복잡한 생체에서 출발해서 단순화를 통해 설명하고 예측하려 해요. 물리학도들은 단순한 이론에서 출발해 복잡한 생체를 설명하고 예측하려 하죠."
공감됩니다. 이토록 복잡한 생체 시스템을 수식이나 이론으로 예측한다는 이야기를 들으면, 생물학도 입장에선 의아할 수 있습니다. 양자컴퓨팅이 그걸 가능하게 해 주는 건가요?
"생체의 모든 현상을 예측하는 건 아직 어렵죠. 다만 기초적인 한 분야에선 가능합니다. 분자 간 상호작용, 바인딩(결합)에 대한 예측입니다.
분자 간 결합을 가능하게 하는 건 결국은 원자 간의 전자 분포예요. 전자 분포가 한 쪽으로 쏠려서 포인트 차지(Point chargeㆍ점전하)를 만들고, 오비탈(Orbital)이 겹치면서 결합력이 생기는 겁니다. 그런데 여기서 일반적인 컴퓨터를 이용한 결합 예측 프로그램이 약점을 보여요."
뭔지 알 것 같습니다. 포인트 차지를 만드는 전자의 분포를 다 계산하지 못하겠네요. 전자는 오비탈 상에서 확정적인 장소에 있는 게 아니라, 확률적으로 분포하니까요. 그 확률을 모두 계산하는 건 일반적인 컴퓨터의 연산능으론 힘들 듯합니다.
"맞습니다. 기존 분자결합 예측 프로그램은 전자의 확률적 분포를 모델링하지 못해요. 원자 한가운데에 포인트 차지가 있다고 가정하니까요.
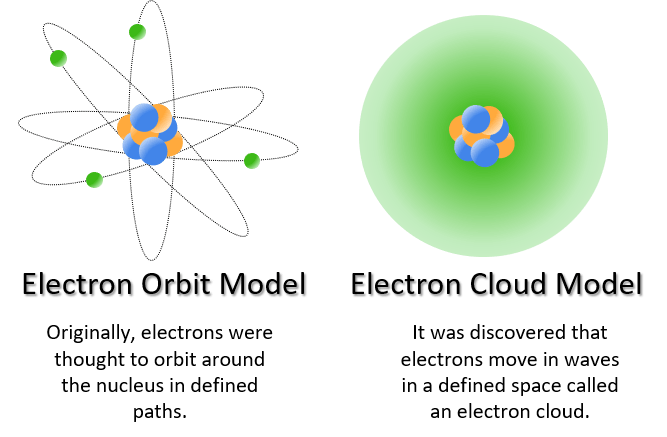
그래서 양자컴퓨팅을 통해 전자 분포와 오비탈을 계산하고, 분자 간 결합을 예측해보자는 것이죠. 다른 복잡한 부분들은 차치하더라도, 최소한 바인딩 포켓(Binding pocket)과 리간드(Ligand)의 결합까지는 볼 수 있도록이요. 리간드 중에서도 크기가 작은 편인 케미컬(Chemicalㆍ저분자 화합물을 의미)까지는 양자 계산을 할 수 있으리라 생각했고, 그러면서 퀀텀인텔리전스의 기반이 되는 프로그램 플랫폼, '퀘스트(QUEST)'를 만들기 시작했습니다."
그 프로그램을 만들기까지 거쳤던 검증 절차가 궁금합니다.
"화학에서 흔히 말하는 큐사(QSARㆍQuantitative Structure Activity Relationshipㆍ정량적 구조 활동 관계) 모델이란 게 있어요. 비슷한 구조의 화합물은 유사한 특성을 가진다는 걸 전제하는 오래된 이론입니다. 즉 화합물의 구조와 결합력, 독성 등은 상관관계가 있다는 것이죠.
이 이론을 바탕으로 화합물의 특성을 예측하게 되는데요. 가령 비슷한 구조를 가진 화합물 A와 B가 있다고 했을 때, A의 특성은 다 알고 있고 B의 특성은 모르는 상태라고 가정해 볼게요. 그럼 둘의 구조가 비슷하니, A의 특성으로 B의 특성을 예측할 수 있겠죠.
이 때 기존 예측 프로그램을 쓰게 되면, 단순한 포인트 차지를 놓고 예측하기 때문에 잘 맞지 않아요. 그런데 전자분포와 오비탈에 대한 양자 계산을 거치니 훨씬 정확한 예측이 가능하더군요."
'더 정확한 예측이 가능했다'는 부분, 자세히 설명해 주세요.
"하나 예를 들자면, 저희가 발표한 논문의 내용인데요. 마우스 실험의 독성 데이터가 있는 1만여 개 화합물 구조를 프로그램에다 머신러닝(Machine learning) 시킵니다. 그리고 특정 화합물의 구조를 가지고 독성 데이터를 예측해 봤어요. 양자역학 이론을 적용해 오비탈 수준에서 계산해 보니, 그 독성 데이터가 실제 실험값과 잘 맞아떨어졌던 거죠. 거기서 가능성을 보고 상업화를 해야겠다는 생각이 들었습니다."
그게 퀀텀인텔리전스의 시작이었군요.
"말 그대로 시작이었죠. 기존에 밝혀진 화합물 구조를 딥러닝시켜서 신규 구조를 예측하는 데에는 한계가 있어서 그렇습니다.
챗지피티(chatGPT)의 퍼포먼스가 좋은 이유는 학습된 데이터가 많아서죠. 그런데 AI 신약개발에선 학습할 데이터 자체가 별로 없어요. 기존 AI 신약개발 사업이 잘 풀리지 않는 이유가 이것입니다.
화합물의 구조를 기반으로 신약이 개발된 사례가 생각보다 많지 않아요. 카이네이즈(Kinaseㆍ인산화효소) 쪽 데이터는 그나마 많은 편이지만요. AI에게 일을 시키려면 화합물 구조에 대한 다양하고 퀄리티 높은 데이터를 학습시켜줘야 하는데, 공부할 자료가 부족한 겁니다.
가령 KRAS만 해도, '소토라십(Sotorasib)'이란 신약이 나오긴 했지만 KRAS 관련한 구조는 몇 개나 나왔나요? 50개 정도 되는 구조 데이터를 AI에 학습시킨다 한들, 새로운 화합물 구조를 창출해낼 수가 없습니다. 재료 없이 요리하라는 것과 마찬가지죠."
그럼 다양한 화합물 데이터를 많이 넣어주면, AI 신약개발이 더 나아질까요?
"잘 구분해서 생각해야 해요. 기본적인 구조가 같은데 유도체가 각기 다른 화합물 10만개를 AI 학습시킨다면, 결국 똑같은 구조에서 유도체만 다른 결과물을 내놓게 돼요. 신규 유도체를 찾는 건 사람이 훨씬 잘하고, 이미 밝혀진 구조의 유도체의 데이터는 매우 많습니다. 빅파마 정도면 다 가지고 있어요.
그래서 기존 AI 신약개발 프로그램을 써서 빅파마에 보여주면, ‘그 화합물 구조는 우리도 가지고 있는데?’라는 반응이 돌아옵니다. 카이네이즈 분야만 해도 이렇고, 다른 분야로 건너가면 이마저도 힘듭니다."
즉 AI 신약개발에선 새 유도체가 아니라 새 구조를 찾아야 의미 있는 것이군요. 그런데 거기엔 재료가 부족하고요.
"그래서 저희는 양자 계산으로 분자결합을 예측하는 알고리즘을 구축하는 데에서 끝내지 않았어요. 그런 알고리즘으로 새롭고 다양한 화합물 구조를 창출해내려면 재료를 대거 넣어줘야죠. 그런 방법으로써 파지 디스플레이(Phage display)를 동원했고요."
파지 디스플레이를 통해서, 타깃 단백질 X에 붙는 다양한 분자구조를 대거 뽑아 '재료'로 쓰는 거네요. 재료를 넣은 다음에 양자컴퓨팅은 어떤 식으로 쓰이나요?
"펩타이드 신약 후보물질을 만든다고 해 볼게요. 한 10mer 정도로 만들었는데, 여기서 2군데 정도만 치환해보고 싶은 거죠. 여기 들어갈 수 있는 비천연 아미노산(Unnatural amino acids)이 3만개 정도니, 가능한 후보물질의 조합이 9억개나 나와요.
여기서 투과도, 용해도, 결합력 등 원하는 물성을 가진 후보물질을 추려야 하는데, 양자컴퓨팅과 AI가 그런 작업을 처리해요. 둘의 역할을 나눠 설명하자면, 양자컴퓨팅은 전자 분포, 구조, 결합 에너지를 계산합니다. AI는 양자컴퓨팅의 결과를 바탕으로 더 적합한 후보군을 좁혀내고요. 두 기술은 상호 보완적으로 작동합니다.
이렇게 나오는 결과를 계속 다시 집어넣는 작업, 즉 피드백 루프(Feedback loop)를 반복시키다 보면, 수백만개에서 시작했던 후보 물질들이 약 30개 정도로 줄어듭니다. 이 정도면 실제 실험으로 테스트할 수 있는 범위가 돼요.
지금 가장 관심도가 높은 분야인 결합력 쪽으로 보면요. 항체의 CDR 리전(region)이 결합력에 관여하고 CDR3가 가장 만지기 좋아요. 여기 들어가는 아미노산 시퀀스가 한 20~30개거든요. 여기서 어디를 건드려야 결합력을 조정할 수 있는지 보고 싶겠죠.
그런데 이 시퀀스를 하나하나 바꿔보면서 실험하는 건 불가능에 가까워요. 시퀀스 하나에 들어가는 아미노산 후보만 20개니까요. 결합력에 관여하는 키스팟(key spot)을 효율적으로 찾는 데에 저희 기술을 쓸 수 있다는 것이죠."
후보물질 발굴에 드는 비용과 인력이 대폭 절감되겠군요.
"기존에 들던 비용의 10분의 1수준, 인력은 5명 정도면 됩니다."
해당 스크리닝 플롯폼이 실제 비즈니스로도 이어지고 있을까요?
"바이오 인터내셔널(BIO InternationalㆍBIO USA)이나 해외 학회를 가면, 머크(Merck), 존슨앤드존슨(Johnson & Johnson), 다케다(Takeda) 같은 빅파마들이 많이 접촉해옵니다. 다만 논의가 진전되는 중에, 그들이 원하는 후보물질 스크리닝을 해 주려면 그들이 가진 인하우스(in house) 데이터가 필요해요. 이걸 제공해달라고 상대 측에 요청하면 난색을 표하는 편이죠.
그러니까 신뢰관계가 형성되지 않았는데 자기 데이터를 넘기는 것이 쉽지 않은 겁니다. 이런 불신의 늪을 뛰어넘으려면 결국 우리 플랫폼으로 성공한 케이스가 나와야 해요. 저희가 자체 신약 파이프라인을 개발하는 이유입니다."
퀀텀인텔리전스의 양자컴퓨팅 플랫폼으로 후보물질의 물성을 예측하고, 예측된 결과가 실제 신약개발에서 재현되는 데이터를 보여주겠다는 것이죠.
"아까 설명한 생물과 물리 간의 벽에 대한 이야기입니다. 이론이 맞으면 결과값도 맞는다는 것을 실제로 보여줘야 벽을 깰 수 있어요."
구체적으로 생각해 두신 전략이 있을까요?
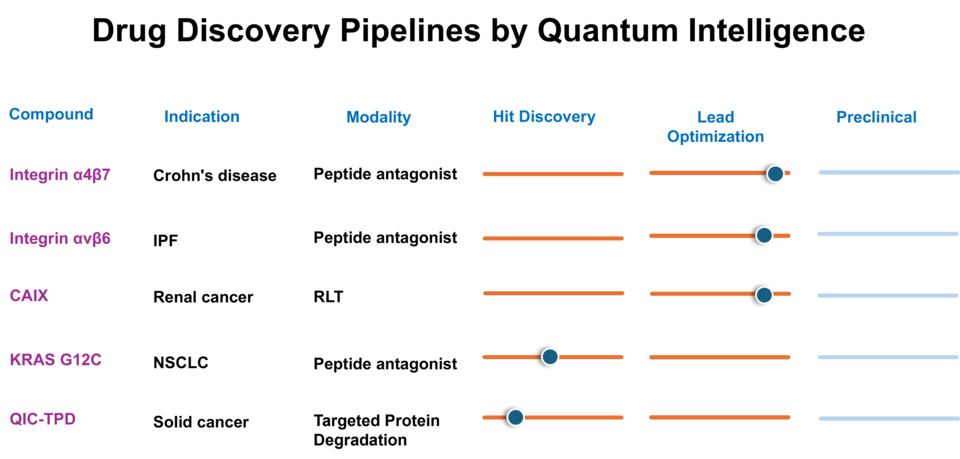
"지금 다케다의 크론병 항체치료제인 베돌리주맙(Vedolizumabㆍ제품명 엔티비오 ENTYVIO)의 특허가 만료돼 갑니다. 그래서 이 항체에서 결합력을 발휘하는 부분을 따다가, 인테그린 α4β7을 타깃하는 펩타이드 후보물질을 개발 중이에요."
항체가 아닌 펩타이드 신약을 개발하는 이유가 있나요?
"항체는 분자의 크기가 너무 커요. 이와 달리 펩타이드는 결합에 실질적으로 관여하는 작은 서열을 중심으로 약물 설계가 가능합니다. 그래서 양자컴퓨팅을 적용해서 후보물질을 더 빠르고 정확하게 도출해낼 수 있어요. 또 지금은 ADC(항체약물접합체)가 관심을 많이 받지만, 그 이후에는 PDC(펩타이드약물접합체)가 부상할 거라고 예상합니다.
마찬가지로 RLT(Radio Ligand Therapyㆍ방사성리간드 의약품) 후보물질도 개발 중입니다. RLT를 개발할 때 중요시되는 부분이 타깃과 리간드 간의 결합을 최적화하는 것이니까요. 리간드 설계ㆍ최적화를 두고 공동개발 혹은 라이선싱을 진행할 수 있으리라 기대하고 있습니다."
요약하자면 퀀텀인텔리전스는 양자컴퓨팅을 통한 후보물질 발굴 플랫폼과 자체 신약개발이란 2가지 사업모델이 있습니다. 자체 신약개발을 통해 플랫폼의 신뢰성을 확보하고, 그 후에 플랫폼 영업을 통해 초기 매출을 낼 수 있겠네요. 자체 신약의 라이선싱도 그 즈음 혹은 후에 이뤄질 수 있겠습니다.
"자체 신약개발에서 적어도 PK(Pharmacokineticsㆍ약동) 데이터를 보여주는 수준, 혹은 임상 1상 IND 파일링(filing)이 가능한 정도의 데이터를 뽑아내면 저희 플랫폼에 대한 신뢰성이 갖춰질 거라 생각합니다.
저희 기술에서 양자컴퓨팅에 대한 부분은 레퍼런스가 강하게 갖춰져 있기도 합니다. 퀀텀인텔리전스는 ‘초격차 스타트업 1000+ 프로젝트’라는 정부 지원사업에서 양자 분야 분야 5개 기업 중 하나로 선정됐어요. 지난 8월에는 IBM의 퀀텀 네트워크 스타트업 멤버(Quantum network startup member)로 합류했고, 미국 큐에라(QuEra), 프랑스 퀀델라(Quandela), 영국 OQC(Oxford Quantum Circuits), 이스라엘 클래식(Classiq) 등 양자컴퓨팅 전문 기업들과 MOU를 체결한 상태이기도 합니다."