
불순물 평가자료 연기할 때 시험 및 등록시기도 의연하게 조정해야
의약품원료 발암 불순물관리는 오리지날 제약회사도 똑같이 어렵다
대한민국 제네릭의약품 정책은 미로에 갇혔다. 어떤 이는 품질 문제를 거론하고, 다른 어떤이는 절대적 품목수를 지목하며, 또다른 어떤이는 위탁제조(CMO) 등 관련 법과 부조화를 지적한다. 히트뉴스는 '발사르탄'을 핵심어 삼아 미래 정책 수립과 시행에 걸림돌이 되는 '불순물' 먼저 걷어내 보려한다. 안개가 걷히면 시야가 선명해 질 수 있다는 믿음으로.
① 제네릭 의약품 정책과 산업 실제 파악의 가림막
② 도랑치고 가재 잡으려는 '1+3 약사법 개정안'
③ 의약품 원료 가격과 NDMA 는 상관성이 있나
NDMA 등 불순물 발생가능성 평가자료 제출기한이 연기될 전망이다. 당초 평가결과는 2020년 5월까지, 시험결과는 2021년 5월까지 식약처에 보고하도록 했었으나, 평가결과 제출기한이 계속 늦어지면서 시험결과 보고기한도 순연되게 되었다.
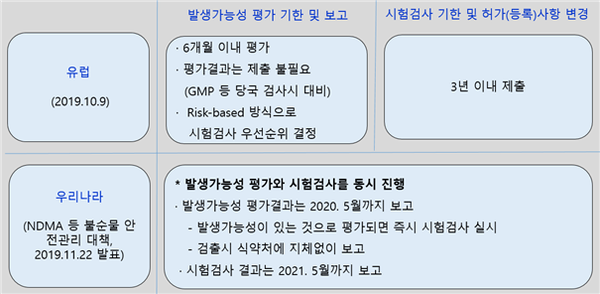
이와 관련해 식약처가 평가기한 연장을 검토할 때 NDMA 사태 당시 불안해 하는 여론을 의식해 평가를 위한 기술적 문제, 한정된 시험검사역량, 원료제조업체와 협력의 문제 등을 충분히 고려하지 않은 일부 무리한 조치사항을 재검토해야 한다는 업계 관계자들의 의견도 여전히 많다.
특히, 발생가능성 평가와 시험검사를 동시 진행하도록 조치한 것은 원료의약품 제조업체에 발생가능성 평가결과를 먼저 받아 봐야 하는 완제의약품 제조업체의 사정상 발생가능성 평가와 시험검사를 동시에 진행하는 것은 무리한 일이었다.
발사르탄 사태 후 미국 FDA가 중국 Huahai사 실사 후 발행한 Warning letter는 "공정 변경을 승인하기 전에 예상치 못한 불순물이 Valsartan API에서 적절하게 검출 및 제어되었는지 확인하기 위한 추가 분석 방법의 필요성을 평가하지 못했다. Huahai사는 제조 공정을 개발하고 변경할 때 불순물을 검출하기위한 적절한 방법을 개발하고 사용할 책임이 있다. 새롭거나 더 높은 수준의 불순물이 검출되면 불순물을 완전히 평가하고 약물이 환자에게 안전한지 확인하기 위한 조치를 취해야 한다"고 지적했다.
Huahai사는 미국FDA 실사 후 현재 산업 관행상 발사르탄 제조 공정 중 NDMA 형성을 예측하는 것이 쉽지 않다고 항변했지만 미국 FDA는 동의하지 않았고, 일반적인 산업 관행이 항상 cGMP 요구 사항과 일치하지 않을 수 있으나 Huahai사가 생산하는 의약품의 품질에 대한 책임은 Huahai사에게 있다고 답변하면서 Warning letter를 발행했다.
미국 FDA의 지적사항과 중국 Huahai사의 항변내용을 보면 현행 규제기준 등으로 의약품원료를 제조하는 데 발생가능한 발암우려물질을 통제하는 것이 쉽지 않다는 것을 보여준다.
이는 라니티딘 사태를 통해서도 알 수 있는 데, 발사르탄의 경우 NDMA는 오리지날제품에서는 검출되지 않았지만 라니티딘의 경우 오리지날 제품에서도 검출되었다. 오리지날 제품이 사용할 것으로 보이는 비싼 원료라고 해서 발암우려물질을 완전히 통제할 수 없는 게 현실인 것이다.
