
NDMA, NDEA, NNV 때문에 의약품 회수 잇따라
정교해진 분석법 맞춰 허가당국과 제조소 대응해야
2018년 고혈압 치료제 발사르탄 원료의약품 불순물 검출을 시작으로, 2019년 라니티딘과 니자티딘 그리고 최근 바레니클린 성분 의약품에서 불순물이 검출되기까지 불순물은 이제 제약산업과 국민건강 측면에서 새로운 리스크로 떠올랐다.
NDMA, NDEA, NNV 등은 N-니트로사민 계열 불순물이라는 공통점이 있다. 식약처와 FDA 등 규제기관들은 불순물 정보를 보도자료나 가이드라인으로 안내하고 있지만, 대중들은 이를 잘 알지 못한다. 언론이 전달하는 내용도 제한적이다. 골치거리로 등장한 니트로사민 불순물 관련 사항들을 정리해 본다. 편집자

2018년 고혈압 치료제인 발사르탄 원료의약품 불순물 검출을 시작으로 3년간 의약품 회수 소식이 이어지고 있으며, 아지도 불순물과 같은 신생 불순물 등장에 대한 걱정과 우려가 이어지고 있다.
발사르탄 원료의약품, 라니티딘 및 니자티딘 성분 의약품 불순물 사태의 주 원인은 ‘NDMA’와 ‘NDEA’였다. 이 물질들은 N-니트로사민 계열 불순물로 구분된다.
ICH(국제의약품규제조화기구)는 ‘DNA 반응성 불순물 평가 및 제어’에 관해 다룬 M7(R1) 가이드라인에서 N-니트로사민 화합물을 ‘우려 집단(Cohort of Concern)’으로 규정하고 있으며, WHO 산하 국제암연구소(IARC)는 ‘그룹2-A(발암가능성이 있는 물질)’로 구분하고 있다.
식약처와 FDA는 가이드라인을 통해 의약품에 존재할 수 있는 N-니트로사민 7가지 종류를 제시했으며, NNV를 포함해 다양한 N-니트로사민 화합물이 존재한다.
FDA와 식약처 기준 설정 닮았지만 그 과정은 달라
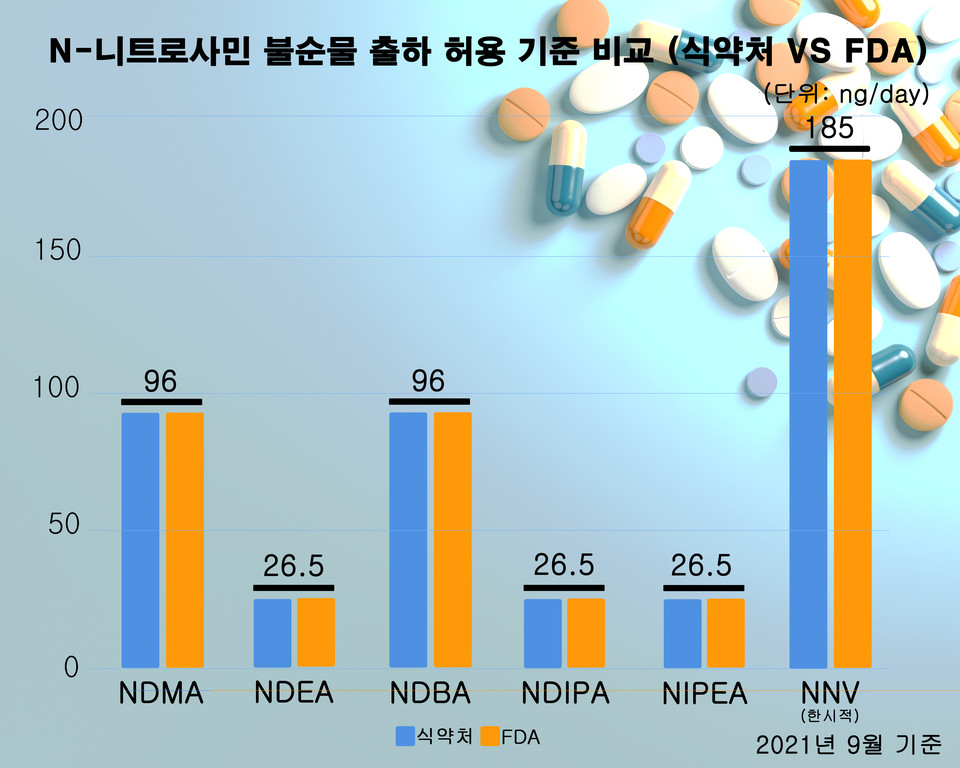
식약처와 FDA는 N-니트로사민 화합물의 일일 허용 섭취량을 다음과 같이 제시하고 있다.
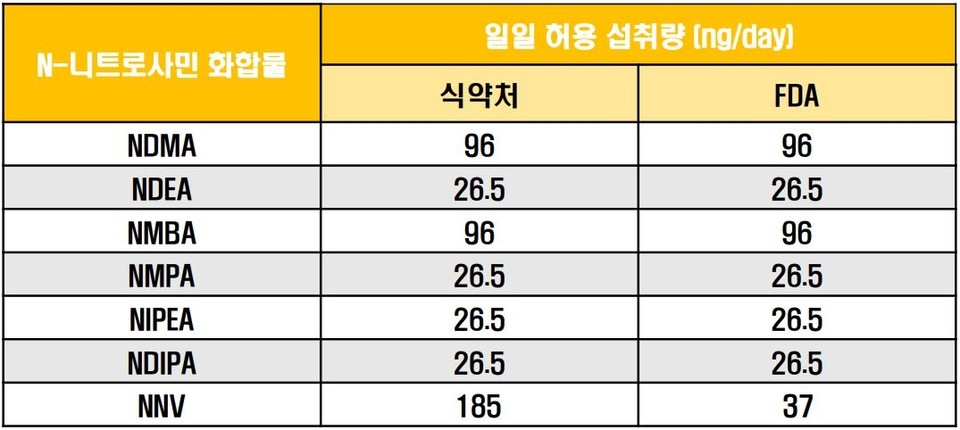
이 기준들은 ICH M7 가이드라인을 통해 0.3ppm 이하 수준으로 설정한 값이다. NNV를 제외하고는 FDA와 동일하다.
식약처와 FDA는 ‘일일 허용 섭취량(ng/day)’을 이 화합물에 70년간 매일 노출될 시 암 발생 위험 증가율 (× 1/100,000)로 정의했다.
식약처와 FDA는 N-니트로사민 화합물을 장기간 섭취할 시 암 발생 위험이 증가할 수 있지만, 일일 허용 섭취 한도 이하로 복용하는 사람은 그렇지 않다고 발표했다.
NNV의 경우는 두 규제기관에서 다른 기준을 설정했다. FDA는 NNV를 함유한 바레니클린 정제를 일일 허용 섭취 한도인 37ng/day 이하로 관리할 것을 제약사에게 권고했고, 유통 수준이 일일 허용 섭취 한도 내로 감소할 때까지 한시적으로 185ng/day로 출하 기준을 설정했다. 반면, 식약처는 중앙약사심의위원회 자문을 통해 FDA의 한시적 출하 기준인 185ng/day로 일일 허용 섭취량과 출하기준을 설정했다.
FDA는 이미 유통 중인 NNV 733ng/day 이하 의약품에 대해서는 회수 대상에 포함하지 않겠다고 밝혔지만, 지난 17일 미국 화이자는 0.5mg, 1.0mg 정제 모든 로트의 자진 회수를 실시하겠다고 밝혔다.
식약처는 현재까지 733ng/day 이하 의약품에 대해서는 자진 회수 대상에 포함하지 않고 있다. 식약처 자문기구인 중앙약사심의위원회에서 △출하 기준 185ng/day를 적용할 시 시중 유통 물량 90% 가량 회수 대상 △화이자 측에서 733ng/day 이상 제품이 전 세계 어디에서도 회수된 적 없다고 주장한 것을 받아들였기 때문이다.
식약처는 화이자의 회수 기준을 따랐지만, 미국에서 모든 로트가 자진 회수되고 있는 상황에서 정작 이 제약사는 국내 회수대상에 포함되지 않았다. 지난 23일 화이자가 회수기준에는 해당하지 않지만 한시적 출하허용기준을 넘는 제조번호에 대해 자진회수를 하겠다고 밝혔다.
N-니트로사민 불순물은 제조과정에서만 생산된다?
미국 USP(미국약전위원회)는 지난해 9월 온라인 세미나에서 "N-니트로사민 화합물이 의약품 제조과정뿐만 아니라 구운 고기, 유제품 및 야채를 포함하여 식품과 물에 일반적으로 포함되어 있다"며, "흡연 시에도 NDMA가 나올 만큼 모든 사람은 일상 속에서 일정 수준의 N-니트로사민 화합물에 노출될 수 있다"고 발표했다.
의약품 제조과정에서 N-니트로사민 화합물은 일반적으로 질소화합물인 2차 아민(Amine)과 아질산염(Nitrite)의 반응에 의해 형성되며, 원인 과정은 '원료의약품 제조단계', '완제의약품 제조단계', '완제의약품 제조 후 단계'로 나눌 수 있다고 설명했다.
원료의약품 제조단계에서 사용된 아민과 아질산염이 정상적으로 제거되지 않은 경우, 완제의약품 제조단계에서 N-니트로사민 화합물을 형성할 수 있으며, 원료의약품이 오염된 경우 완제의약품에서 N-니트로사민 화합물이 형성에 직접적인 원인이 될 수 있다고 주장했다. 식약처는 과거 발사르탄 원료의약품 불순물 사태는 이 경우로 추정한 바 있다.
완제의약품 제조단계에서 온도, pH 또는 시약, 용매의 추가 순서와 같은 반응 조건이 부적절하거나 제대로 제어되지 않는 경우 N-니트로사민 화합물을 형성할 수 있으며, 후속 공정으로 넘어가면서 또는 오염된 용수(Water)나 용매의 사용이 원인이 될 수도 있다고 설명했다.
완제 의약품 제조 후 단계에서 세척 과정, 보관 중 제품 자체 분해, 포장 물질 등에 의해 N-니트로사민 불순물이 발생할 수 있다고 덧붙였다.
이 원인들은 동일 원료의약품을 사용한 공정 과정 내에서도 발생 양상이 다를 수 있으며, 어느 단계에서 발생했을 지 추정할 뿐 확실한 원인을 찾기는 힘들다는 것이 규제기관들의 주장이다.
빈번해진 의약품 불순물 검출, 사전 대비 가능한 분석법 필요
기존 미국을 비롯한 주요 선진국에서 정상적으로 품목허가를 받아 판매되어 온 의약품들에서 새로운 불순물들이 검출되고 있다.
한 원료의약품 품질보증 실무자는 "과학기술의 발전으로 불순물 분석법은 점점 정확해지고, 민감해지고 있는 것이 원인 중 하나일 수 있다"며, "극소량의 불순물을 검출해야 하는 만큼 분석법에 따라 검출 범위가 달라질 수 있다"고 주장했다.
식약처와 FDA는 N-니트로사민 화합물을 검출하기 위해 제약회사가 선택할 수 있는 분석법을 웹사이트에 게시해 놓았다. 각 분석법은 검출 불순물 종류와 민감도가 다르며, 제약회사는 자사 분석법을 개발하여 사용할 수도 있다. 이 경우 규제기관은 불순물 검출 수준 검증을 요구하고 있다.
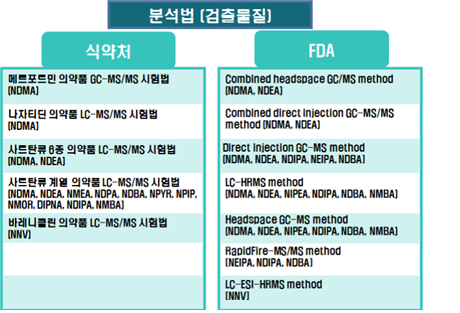
규제기관은 원료의약품 및 완제의약품 제조업체에게 N-니트로사민 화합물의 일일 허용 섭취량을 충족하기 위해 0.03ppm(part-per-million, 100만분율) 이하 수준의 정량 한계(LOQ)를 가진 분석법을 요구하고 있다.
업계관계자는 “의약품 특성에 맞는 분석법 개발과 시설 구축은 제조업체에게 부담이기 때문에 식약처에서 국제 규격에 조화하며, 불순물 이슈에 사전 대비 가능한 다양한 분석법을 제공해줬으면 한다”고 덧붙였다.
불순물 리스크, 끝이 아니다
예상치 못한 의약품 불순물 검출에 각 국 규제기관은 대안을 세우고, 조치를 취하고 있다. 미국과 한국은 주요 선진국 규제기관과 상의 및 ICH 가이드라인을 참고하여 기준을 설정하고 있다는 점에서 동일하지만, NNV 일일 섭취 허용량과 회수 기준 설정 과정을 보면 FDA가 더 보수적으로 접근하고 있다.
식약처는 의약품 제조과정에서 불순물 생성 혹은 유입으로 인한 문제를 예방하기 위해 여러 대책을 제시하고 있다고 밝혔다.
불순물 사태 발생 후 제조업체 자체점검을 통해 불순물이 발생할 수 있는 의약품의 발생 가능성 평가와 시험검사를 실시해 보고하도록 했으며, 해외 제조소를 통해 제조하는 경우 식약처에 해외 제조소 등록을 통해 GMP 기준 준수 확인을 위한 서류 제출 및 제조시설 실사를 의무화하는 '해외 제조소 등록 및 현지 실사 제도'를 시행하고 있다.
식약처는 현재보다 더욱 능동적으로 대처해 의약품 관련 불순물 이슈로부터 제약업계가 신속하게 대응할 수 있도록 지원하며, 안전관리 책임기관으로서 국민들의 불안감을 해소하는데 주력하겠다는 입장이다.
참고자료
1. ICH 가이드라인 M7(R1) ‘Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk’
2. FDA 가이드라인 ‘Control of Nitrosamine Impurities in Human Drugs’
3. USP 2020년 9월 웨비나 발표자료 ‘Nitrosamine impurities An Overview’