
50개 이상 신약 통과 '사상 최고치' 근접
신속심사가 가져올 잠재적 이슈는 뭔가
지현배 박사의 샌디에이고 통신 [9]
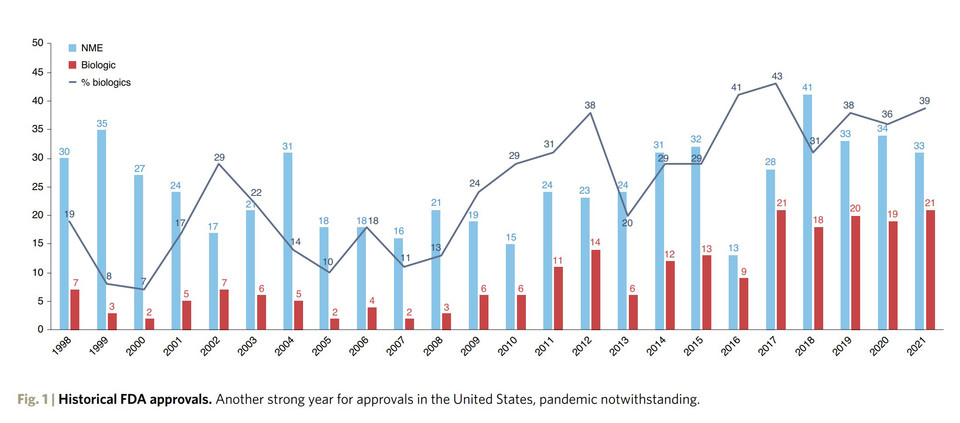
2021년에는 미국 FDA가 약물의 승인을 가속화하고 COVID-19를 대처하기 위한 지속적인 노력이 있었지만 일부 제약 회사에게 너무 유리하게 바뀐 현상이 관찰되고 있다. 50개 이상의 신약이 통과되었는데 이는 2018년의 사상 최고치에 가까우며 첫 번째 팬데믹 연도인 2020년과 동등한 수준이다(그림 1 및 표 1). FDA는 평소의 업무량 외에도 COVID-19 백신 및 치료제에 대해 이미 신속한 승인 일정을 가속화해야 한다는 정치적 압력에 직면했다.
FDA 의약품 자금의 거의 3분의 2가 현재 업계 사용자 수수료로 제공됨에 따라 신속한 검토가 표준이 되고 있다. 그러나 기관 및 기타 규제 기관이 검토를 신속하게 진행에 따라 약물에 대한 심각한 안전성 문제가 발생할 수 있다. 이러한 약물에 대한 위험성이 감지되기 전에 향후 비-COVID-19 관련 약물의 승인 일정을 얼마나 앞당길 수 있을까? 그리고 파이프라인과 승인이 모두 틈새 질환에 치우쳐 있기 때문에 현재 개발 인센티브가 대부분의 환자에게 영향을 미치는 만성 질환을 해결하는 데 적절한 것일까? 이러한 질문들을 통해 향후 신속한 검토가 가져올 수 있는 잠재적인 이슈를 살펴봄으로 궁극적으로 국내 바이오 기업들이 신약 개발 승인을 위한 전략을 세우는데 도움이 되고자 한다.
힘겨웠던 또 한 해
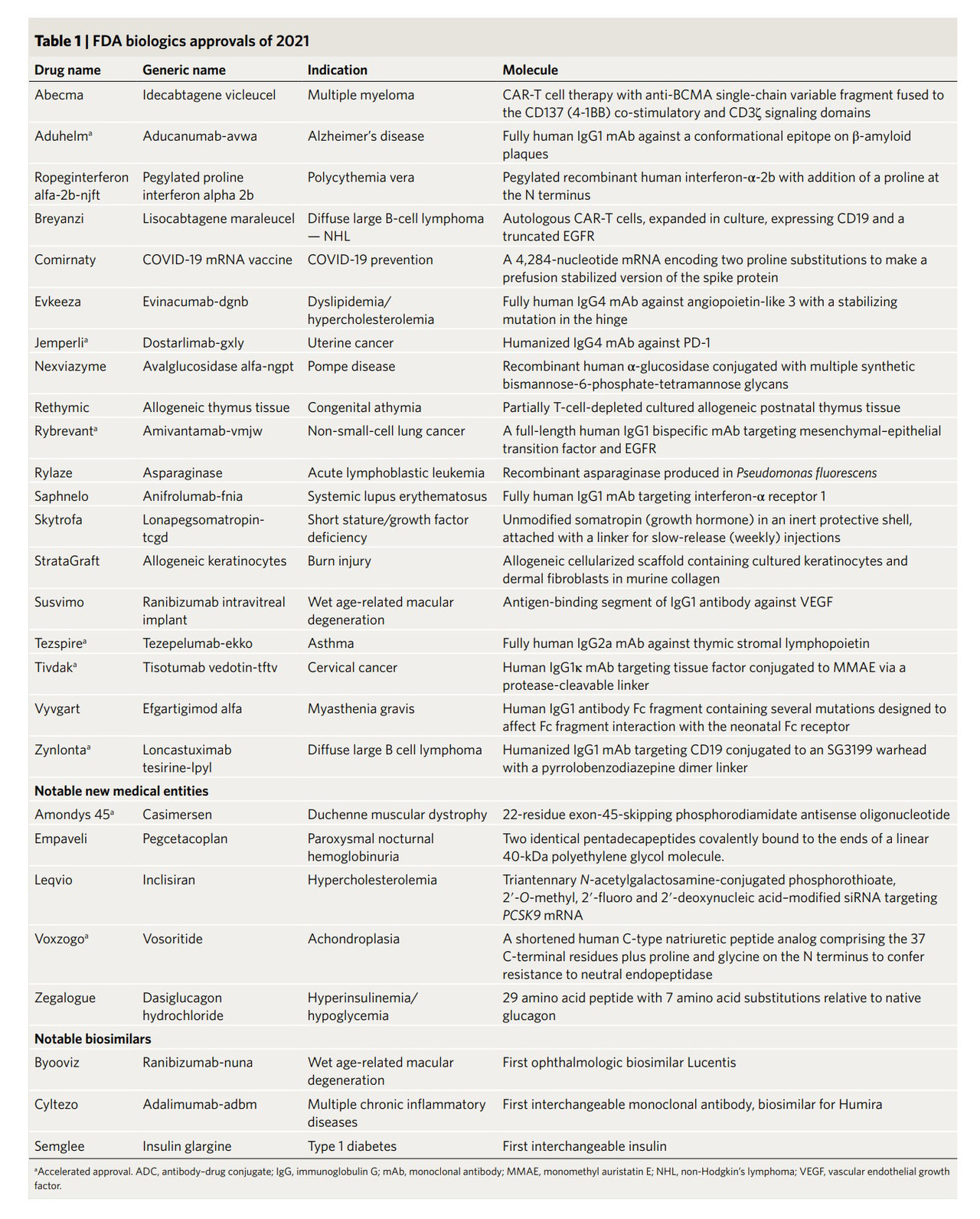
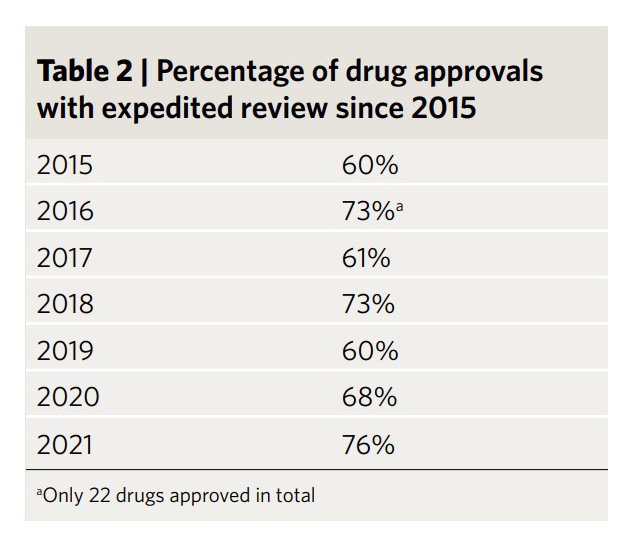
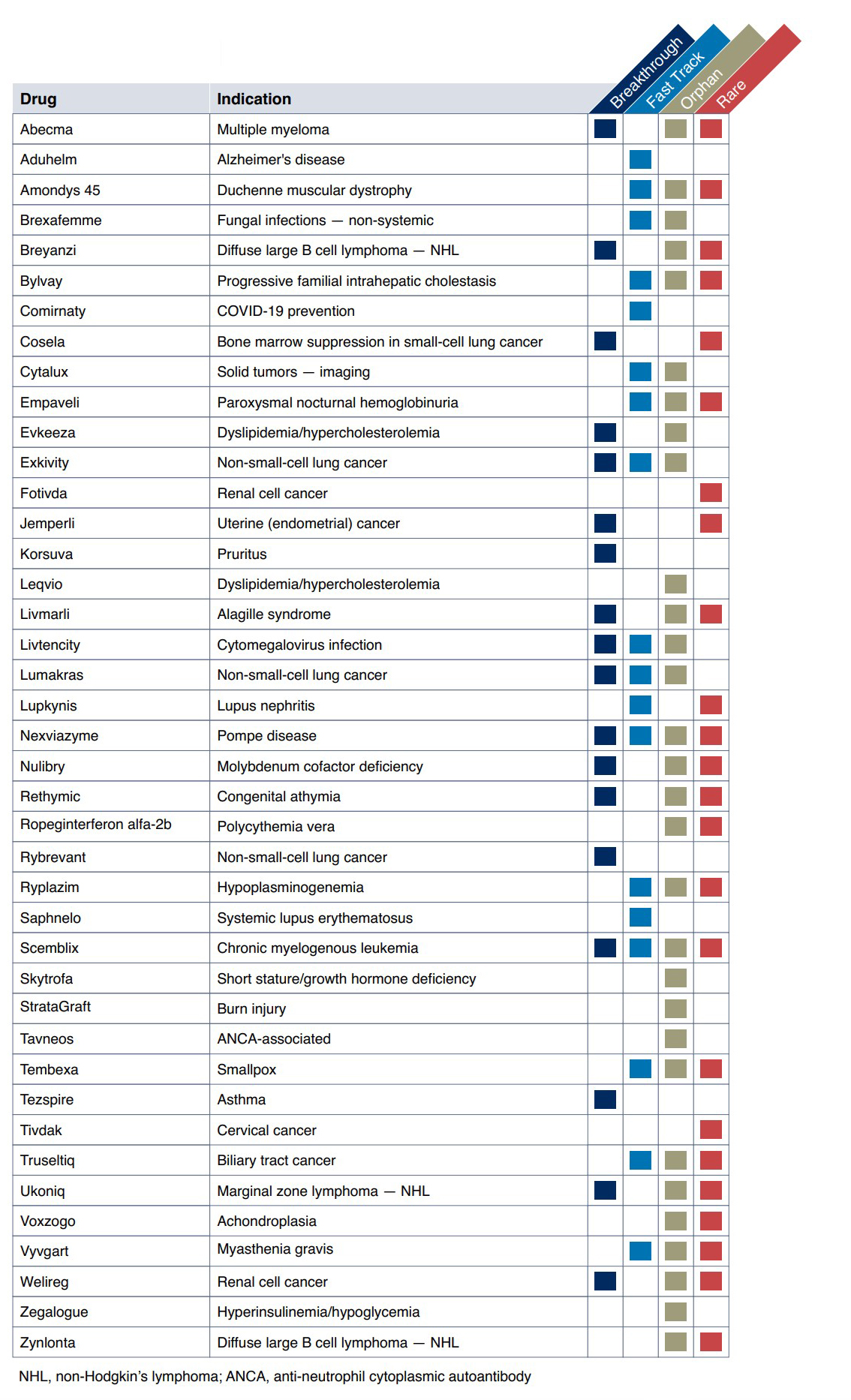
연간 FDA 약물 승인은 15년 전보다 두 배 증가했다. 이는 과학적 진보의 신호일 뿐만 아니라 업계가 규제 기관과 그 어느 때보다 긴밀한 관계를 맺고 있다는 신호이기도 하다. 미충족 분야에서 새로운 치료 옵션에 다양한 형태로 적용되는 신속 승인은 새로운 표준이 되고 있다. 이 신속 승인된 약물은 승인된 모든 신약의 약 70%를 차지했다(표 2).
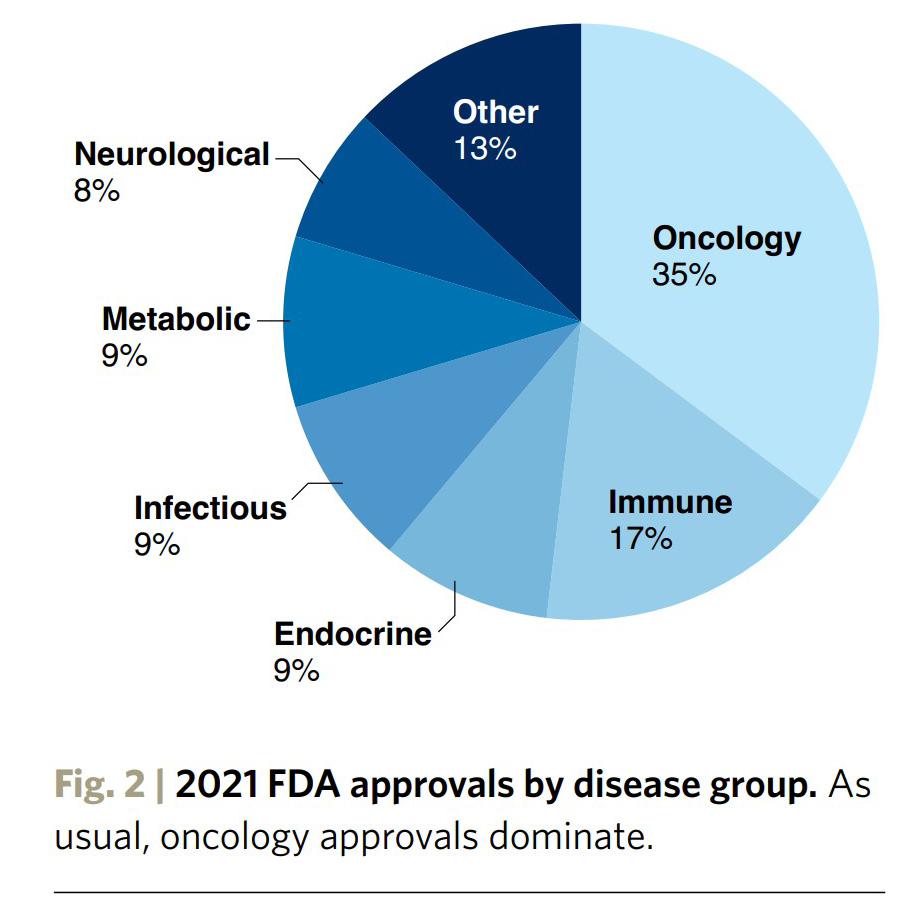
종양 약물이 신속 및 정기 승인 명단에서 다시금 우세했다 (그림 2). 미국의 바이오 제약 의약품 규제, 정책 및 시장 접근을 분석하는 Prevision Policy의 Michael McCaughan에 따르면 모든 종양학 리뷰를 감독하는 준 자율 단위인 FDA의 Oncology Center of Excellence(OCE)는 팬더믹 상황에도 불구하고 "암 환자에게 여전히 중요하다는 메시지를 보내기 위해 의식적인 노력을 기울였습니다"고 설명했다. 다른 규제 기관에 따르면 20건의 항암제 승인 중 8건은 FDA가 관여한 Orbis 프로젝트를 통해 다른 국가에서 동시에 제출 및 검토되었다 (그림 3).
팬데믹이 2021년까지 계속되는 동안 화이자와 바이오엔텍의 mRNA 기반 코로나19 백신 코미르나티는 최초로 긴급사용승인(Emergency Use Authorization, EUA)을 거쳐 완전한 승인을 받았다. 올해 마지막 몇 주 동안 돌연변이가 심한 백신 회피 오미크론 변종이 확산되기 시작하면서 젊은 연령층을 포함하여 화이자 및 모더나 백신의 추가 접종을 위해 EUA가 발급한 것이다. 또한 여러 COVID-19 치료제가 있다. 가장 주목할만한 것은 12월 22일에 화이자의 Paxlovid (nirmatrelvir 와 ritonavir)가 진행 위험이 있는 경증 내지 중등도의 COVID-19 환자에 대한 응급 승인을 받은 최초의 경구용 COVID-19 치료제가 된 점이다. Paxlovid는 입원 또는 사망 위험을 89% 줄이고 복용하기 쉽기 때문에 특히 미국 인구의 20% 이상이 아직 예방 접종을 받지 않은 상태에서 바이러스 퇴치에 중요한 치료용 약물이다. 5일 동안 1일 2회 투여하는 또 다른 요법은 바이러스 복제를 차단하는 프로테아제 억제제인 nirmatrelvir의 분해를 늦춰 억제 효과를 연장시키는 ritonavir와 병용하는 요법이다.
다른 약물 규제 기관과 마찬가지로 FDA는 새로운 작업 패턴, 원격 기술 및 새로운 수준의 국제 협력에 적응하면서 팬데믹을 통해 놀라운 민첩성과 생산성을 보여주었다. 그러나 무대 뒤에서의 상황은 그리 순조롭지만은 않았다. 이 기관은 직원의 피로, 경영진의 혼란, 가속화된 승인 경로의 느슨한 기준에 대한 비판에 직면해 있다. 또한 FDA는 거의 1년 동안 상임 리더가 없었고, 일부에서는 예측할 수 없는 결정을 내렸다고 알려져 있다. 정책 연구 그룹 Capital Alpha의 의료 분석가인 Rob Smith는 "리더십 공백이 있을 때 직원의 권한이 더 커지고 특정 개인이 결정을 내릴 가능성이 더 높아집니다."라고 설명했다.
예측할 수 없는 결정에 대한 좋은 예는 부정적인 자문위원회 투표에도 불구하고 바이오젠의 알츠하이머 치료제 아두헬름(아두카누맙)이 6월에 승인을 받은 것이다. Aduhelm은 알츠하이머, β-아밀로이드 플라크와 관련된 근본적인 병리생리학의 일부를 치료하기 위해 승인된 최초의 약물이다. 그러나 β-아밀로이드가 대리 평가 변수로서의 관련성, 환자당 연간 56,000달러의 가격(12월에 절반으로 인하), 폭넓은 라벨(한 달 후, 모든 알츠하이머병 환자로부터 경증 환자로 축소됨)에 대해 혹독한 비판이 있었고 확인 증거를 제공하는 조건으로 Biogen에게 9년이 부여되었다. FDA 직원 간의 논쟁, FDA의 최고 알츠하이머병 규제 기관의 Billy Dunn과 Biogen 간의 지나치게 친밀한 관계에 대한 주장, 가속화된 승인의 오용에 대해 미국 보건복지부 감찰국에서 조사가 진행 중이다. 또한 유럽의 인체용 의약품 위원회(Committee for Medicinal Products for Human Use, CHMP)는 2021년 마지막 날에 이 약의 승인 요청을 부결시켰다.
미국 메디케어 및 메디케이드 서비스 센터(Centers for Medicare and Medicaid Services, CMS)는 약품 보장을 비정상적으로 연기했으며 2022년 1월에는 적격 임상 시험에 참여하는 환자에게만 비용을 지불할 것을 제안한 상황이다. FDA 커미셔너인 Robert Califf가 지휘를 맡은 것과 마찬가지로 최종 보장 결정과 감찰관의 결정은 올해 말 예정이다. 그는 2021년 10월에 가속화된 검토를 위해 제출된 Eli Lilly의 β-아밀로이드를 표적으로 하는 또 다른 치매에 대한 단일클론항체(mAb) 약물인 donanemab를 처리하는 방법을 포함한 많은 요구에 직면하게 될 것으로 예상된다. 이 약물 또한 한계 효능 결과를 보고했으며 일부 임상 결과를 충족하지 못했다.
대부분의 FDA 관계자들은 2016년 위원으로 이전 FDA 경험이 있는 심장 전문의이자 임상 시험의인 Califf를 환영하고 있다. 어떤 사람들은 그가 최근에 Verily Life Sciences의 수석 고문으로 일하면서 업계와의 관계에 의문을 제기했지만 그는 이런 문제에서 이전 사람과는 거리가 먼 것으로 알려져 있다. Califf는 이미 관료주의를 줄이고 데이터 투명성과 상호 운용성을 개선하여 임상 시험을 간소화할 계획을 선언한 상태이다.
이러한 변화는 신약 개발을 더욱 가속화하여 환자에게 더 빠른 접근을 제공하고 산업에 도움이 될 것으로 기대된다. 또한 2021년에 발표된 FDA의 2023-2027년 목표에는 비종양 적응증에 대한 보다 신속한 검토와 직원 증가가 포함되었다. FDA가 개별 환자의 특정 유전 질환 변이를 위해 개발한 'n-of-1' 치료법에 대한 지침을 발표함에 따라 초희귀 질병 (super-rare diseases)에 대한 맞춤 의약품도 2021년에 증가했다. 절차상의 조언은 ‘n-of-1’ 치료의 가장 진보된 범주인 안티센스 올리고뉴클레오티드 약물과 관련이 있지만 그 이상으로 확장될 가능성이 있다.
COVID-19 비상 사태는 새로운 적응증 또는 승인 후 요구 사항을 지원하기 위해 실제 증거를 수용하려는 FDA의 노력에 탄력이 되었다. 2021년에는 전자 건강 기록, 의료 청구 데이터, 모바일 장치 및 데이터 표준, 데이터 품질, 선택 및 분석에 대한 일반 지침을 포함하는 등록 데이터 사용을 다루는 3가지 지침 초안이 2021년에 발표되었다. FDA는 여전히 실제 증거를 기존의 중재 시험을 대체하기보다는 보조적인 것으로 보고 있다. 그러나 2023-2027년에 계획된 지침 및 추가 노력의 양과 전통적인 연구가 어려운 희귀 질환 치료제로의 전환에서 실제적인 증거를 확보하는 것은 더 중요해질 것으로 전망된다.
쏟아지는 신약들
2021년의 승인은 새로운 목표를 달성하거나 질병 아형 또는 암 돌연변이에 대한 첫 번째 치료 옵션을 제공하는 약물로 가득 차 있다.
신약 개발 분야에서 가장 흥미로운 승인은 이전에 약물 개발이 불가능하다고 믿었던 표적을 공격하는 Amgen의 저분자 약물인 Lumakras(sotorasib)이다. Lumakras는 많은 암과 관련된 활성 GTP 결합 및 비활성 GDP 결합 구조 사이를 전환하는 GTPase인 Kirsten rat sarcoma (KRAS) 단백질의 G12C 돌연변이를 억제한다. KRAS G12C는 비소세포폐암(NSCLC)에서 가장 흔한 돌연변이 아형이다. Lumakras는 KRAS의 유도성 알로스테릭 스위치 포켓 근처에 Cys12 잔기를 공유 결합함으로 작동하여 세포 증식을 멈추는 비활성 상태로 고정시킨다.
신약 승인은 프로젝트 Orbis를 통해 호주, 브라질, 캐나다 및 영국의 당국과 조정되었다. 이는 규제 기관 간의 더 큰 국제적 협력뿐만 아니라 과학적으로 약물 표적에 접근하는 공유 분자 및 알로스테리의 부활을 보여주고 있다. 많은 승인된 약물(예: 아스피린 및 페니실린)이 공유 결합하지만, 공유 결합의 비가역적 특성은 맨틀 세포 림프종에 대한 Janssen의 Imbruvica(ibrutinib)가 2013년 승인될 때까지 주류 약물 설계에서 밀려났었다. 이 약물들의 개발은 단백질 모형을 더 자세히 시각화하여 이해할 수 있는 새로운 접근법을 통해 가능했다. Bruton’s tyrosine kinase 의 저분자 억제제인 imbruvica는 이후 베링거인겔하임의 Giltorif (afatinib), 아스트라제네카의 Tagrisso (osimertinib) 와 Calquence (acalabrutinib) 같은 다른 공유결합 억제제의 르네상스를 촉발했다.
유전적으로 정의된 질병에 대한 승인은 강력한 속도를 유지했으며, 처음 2 건은 표피 성장 인자 수용체(epidermal growth factor receptor, EGFR) 엑손 20에서 돌연변이된 NSCLC 환자 치료용이다. Janssen의 이중특이성 mAb Rybrevant(amivantamab-vmjw)와 Takeda의 경구용 저분자 선택적 키나제 억제제인 Exkivity(mobocertinib)는 모두 백금 기반 화학 요법 후 질병이 진행된 사람들을 위해 가속화된 승인을 받았다. NSCLC의 약 2-3%가 이런 돌연변이를 가지고 있다.
Duchenne의 근이영양증 (Duchenne’s muscular dystrophy, DMD)과 같은 희귀 질병도 계속해서 더욱 세분화되고 있다. 2021년에는 엑손 45 건너뛰기가 가능한 디스트로핀 (dystrophin) 유전자 돌연변이가 있는 환자의 8%를 위한 phosphorodiamidate morpholino 안티센스 올리고뉴클레오티드인 Sarepta의 Amondys 45(casimersen)가 FDA 승인을 받았다. 회사의 다른 두 가지 DMD 약물인 Exondys 51(eteplirsen)과 Vyondys 53(golodirsen)은 각각 엑손 51 및 53이 결손된 소수의 환자를 대상으로 하고 있다. 치료는 더 짧은 dystrophin 단백질의 발현을 촉진하지만 dystrophin 생산 증가가 임상적 이점으로 이어질 것이라는 논란의 여지가 있는 가정 하에 모두 가속화된 방식으로 승인되었습니다. 이러한 점은 확증적 시험에서 확인될 필요성이 있다.
보체 시스템 단백질과 효능에 대한 이해가 높아짐에 따라 염증 및 면역 관련 질병에 대한 프로그램이 추진되고 있다. ChemoCentryx의 Tavneos(아바코판)는 심각한 항호중구 세포질 자가항체(severe active anti-neutrophil cytoplasmic autoantibody, ANCA) 관련 혈관염, 희귀 염증성 질환 치료를 위해 보체 인자 5a(C5a) 수용체를 선택적으로 억제하는 저분자 약물로서 미국과 일본에서 승인을 받았으며 중증 활동성 환자에서 표준 글루코코르티코이드 요법에 대한 추가 치료제로 유럽 연합에서 긍정적인 평가를 받았다. Omeros의 완전 인간 면역글로불린 G4(IgG4) 단일항체 치료제인 narsoplimab은 면역글로불린 A 신병증 (immunoglobulin A nephropathy, 신장 염증)에 대해 임상 3상 단계에 있다. 이 항체 치료제는 렉틴 활성화 경로(세 가지 보체 활성화 경로 중 하나)의 작용 효소인 MASP-2 (mannan-binding lectin-associated serine protease-2)를 표적으로 하며 미국과 유럽 연합에서 희귀 약물 지정을 받았다.
Apellis의 펩타이드 약물 Empaveli(pegcetacoplan)는 2021년 5월 결함이 있는 적혈구가 면역 체계에 의해 공격받는 희귀 혈액 질환인 발작성 야간 혈색소뇨증 (paroxysmal nocturnal hemoglobinuria)에 대해 최초로 승인된 C3 조절제가 되었다. Empaveli는 2007년 초기 발작성 야간 혈색소뇨증 적응증에서 승인된 C5-Soliris(eculizumab)를 표적으로 하는 Alexion의 mAbs보다 헤모글로빈 증가에 더 큰 효능을 보여준 바가 있다. Ultomiris(라불리주맙 cwvz)은 2018년에 승인 받았다. 이런 결과는 C3가 하위단계의 C5 단백질보다 면역계의 보체 연속 반응에서 더 중심적이기 때문이다. 즉 C3는 혈관외 및 혈관내 용혈을 모두 제어하는 반면 C5는 혈관내 용혈에만 관련되어 있다.
11월에는 또 다른 펩티드 약물인 BioMarin의 Voxzogo(vosoritide)가 가장 흔한 왜소증 질병인 연골무형성증 (achondroplasia)에 대해 승인된 최초의 약물이 되었다(유럽 및 영국 승인도 받음). 연골무형성증이 있는 사람들은 뼈 형성을 억제하는 과활성 섬유아세포 성장 인자 수용체 3(FGFR3) 유전자를 가지고 있다. C형 나트륨 이뇨 펩타이드의 유사체인 Voxzogo는 나트륨 이뇨 펩타이드 수용체 B (natriuretic peptide receptor B)에 결합하여 FGFR3의 효과를 약화시켜 골 성장을 자극한다. Voxzogo의 가속화된 승인에는 최종 성인 키와 청력 상실, 수면 무호흡증 및 골격 문제를 포함하여 연골무형성증으로 인해 발생할 수 있는 임상 문제에 대한 확인 데이터가 필요한 상황이다.
연골무형성증은 미국에서 단지 10,000명의 어린이에게 영향을 미치지만 Voxzogo는 오랫동안 유일한 치료법이 아닐 수 있다. 또한 옹호 단체인 Little People of America를 비롯한 일부에서는 주로 신장에 초점을 맞춘 약물이 필요한지 의문을 제기하고 있는 실정이다. 다른 제약회사들은 비정상 단백질이 매개하는 연속 반응에 초점을 맞추고 있다. Ascendis Pharma는 연골무형성증에 대한 2상에서 TransCon CNP 단백질의 주 1회 제형으로 뼈 시뮬레이션 C형 나트륨 이뇨 펩티드를 지속적으로 주입하는 것을 목표로 하고 있다. 화이자의 FGFR3 미끼인 리시퍼셉트(recifercept)도 임상 2단계에 있다. Recifercept는 FGFR3 리간드를 격리하여 돌연변이된 수용체의 활성화를 감소시킨다. BridgeBio Pharma의 임상 3상 시도에 있는 infigratinib은 FGFR1-3의 선택적 경구 티로신 키나제 억제제로, 2021년 5월 FGFR2 융합 또는 재배열이 있는 진행성 또는 전이성 담관암 환자를 위한 Truseltiq으로 승인되었다.
다른 형태의 전염병
2021년 처음으로 승인된 약물들은 더 광범위한 건강 분야에 영향을 미쳤다. 2021년 6월 Novo Nordisk의 Wegovy(semaglutide 2.4mg)는 식이요법 및 운동과 함께 평균 15%의 체중 감소를 보여주며 비만 수술과 동등한 효능을 나타내는 최초의 FDA 승인 비만 약물이 되었다. Semaglutide 는 새로운 형태의 약물이 아니다. 글루카곤 유사 펩티드(glucagon-like peptide, GLP)-1 유사체는 2017년부터 당뇨병에서 더 낮은 용량으로 사용되었다. GLP-1 약물은 2005년부터 사용 가능했다. 비만 전문가들이 매우 흥분하는 이유는 Wegovy가 다른 많은 항비만 약물과 달리 매우 높은 안전성을 보여주며 효능을 보이는 약물이기 때문이다.
코로나바이러스 대유행은 미국에서만 1억 명이 넘는 비만 환자들이 직면한 추가적인 위험 요소가 되었기 때문에 출시 후 약물구입의 양이 증가함에 따라 부분적으로 약물 공급 부족의 원인이 되었다. 비만인 사람들에게서 볼 수 있는 염증 및 면역 체계 손상은 연령 및 관련 상태와 같은 다른 요인과 상관없이 심각한 COVID-19 증세, 입원 및 사망 위험을 증가시키는 것으로 나타났다. 최근 미공개 연구에 따르면 지방 조직 자체가 SARS-CoV-2 침투 및 관련 염증에 취약하며 바이러스 복제를 위한 저장소를 제공할 수 있다고 보고되었다. Weill Cornell Medicine의 종합 체중 조절 센터의 대사 연구 교수인 Louis Aronne은 "팬데믹은 마침내 모든 사람(임금 지급인, 고용주, 대중)에게 비만이 심장병, 당뇨병, 비알코올성 지방간염 및 우울증을 포함한 건강 위험과 관련된 질병이라는 것을 확신하게 했습니다", "그리고 이제 우리는 이전 제품보다 두 배 이상 효과가 좋은 Wegovy라는 약을 갖게 되었습니다." 라고 평가했다.
CMS는 법률에 따라 고혈압이나 당뇨병과 같은 관련 질환에 대한 치료의 일부가 아닌 한 항비만 약물에 대해 비용을 상환하지 않는다. 그러나 Novo Nordisk는 상업 시장에서 60%의 적용 범위를 달성했으며 Wegovy은 3대 약국 혜택 관리자가 제공하는 플랜에 포함되어 있다고 말했다. 애널리스트의 매출 예측 범위는 40억 달러에서 90억 달러이다. 이 약은 유럽의 CHMP에서도 긍정적인 평가를 받았고 일본에서도 검토 중이다.
Novo Nordisk만이 이러한 서비스가 부족한 시장을 공략하려는 것은 아니다. Novo Nordisk은 2형 당뇨병에 대한 FDA 검토와 비만에 대한 3상 시험에서 매주 1회 이중 포도당 의존성 인슐린 분비 촉진 폴리펩타이드(glucose-dependent insulinotropic polypeptide, GIP) 및 GLP-1 수용체 작용제인 일라이 릴리의 tirzepatide와의 경쟁에 직면해 있다. GLP-1과 GIP 펩타이드 작용제를 결합하면 GIP의 독립형 효과 중 일부가 역효과(예를 들어 지방 저장을 증가시키는 것으로 보이는 현상)를 내는 것처럼 보이지만 동시에 부가적인 것으로 보일 수 있다. Tirzepatide는 예상되는 미래 판매의 순 현재 가치 측면에서 업계에서 가장 가치 있는 후기 개발 후보 중 하나이다. 두 약물은 또한 심부전 및 비알코올성 지방간염에서 테스트되고 있다.
2021년에 바이엘 (Bayer)은 심부전과 신장 질환이라는 널리 퍼진 다른 두 가지 만성 질환에서 더 많은 옵션을 제공했다. Merck와 공동 개발한 Verquvo(vericiguat)는 1일 1회 경구용 가용성 guanylate cyclase 자극제로서 산화질소 신호 전달 경로를 통해 평활근 이완을 유발하여 심부전의 내피 기능 장애를 개선하는 약물이다. 박출률이 45% 미만인 심부전 악화에 대한 표준 치료의 추가 기능으로 승인되었다. 5,000명의 환자를 대상으로 한 시험에서 심혈관 원인 및 관련 입원으로 인한 사망의 복합 위험이 10% 감소하는 것으로 나타났지만 심혈관 관련 사망의 감소만으로는 의미가 없었다.
비스테로이드성 무기질코르티코이드 수용체 길항제 Kerendia (finerenone)는 만성 신장 질환을 갖고 있는 제2형 당뇨병 성인의 35% 이상을 대상으로 한다. 이 약물은 신부전과 심부전을 유발할 수 있는 사구체 여과율의 감소를 늦추고 둘 다의 위험을 줄이는 것으로 나타났다.
2021년 승인된 암 이외의 큰 질병에 대한 저분자의 두 약물은 당뇨병에 사용되는 기존 SGLT-2 억제제가 갖고 있는 문제에 직면해 있다. AstraZeneca의 Farxiga(dapagliflozin)도 당뇨병이 없는 환자의 신장 질환에 대해 승인되었으며, 이 계열은 이 광범위한 그룹에서도 심장 보호 효과를 보여주었으며 잠재적으로 혈압을 낮추고 심장 섬유증과 내피 기능 장애를 모두 억제하는 등 여러 기전에 의해 매개된다.
제조 현장 관련 거부(및 EU 승인) 1년 후인 2021년 마지막 날, FDA는 Novartis의 콜레스테롤 저하제 Leqvio(inclisiran)를 허가했다. 간에서 proprotein convertase subtilisin-kexin type 9 (PCSK9)의 발현을 차단하는 이중 가닥 소형 간섭 RNA 약물은 식이 요법 및 스타틴과 함께 죽상 경화성 심혈관 질환을 앓고 있는 1,800 만 명 정도의 미국인과 이형 가족성 고콜레스테롤혈증 (heterozygous familial hypercholesterolemia) 환자들을 치료하기 위해 승인되었다. Leqvio는 저밀도 지단백 콜레스테롤 (low-density lipoprotein cholesterol)을 감소시키고 1년에 2회 피하 주사한다.
바이오시밀러와 단일 항체 시장
2021년에 승인된 두 개의 바이오시밀러도 잠재적으로 공중 보건에 큰 영향을 미칠 수 있다. Biocon의 Semglee (insulin glargine -yfgn)는 Sanofi의 Lantus의 바이오시밀러이고 베링거인겔하임의 항체 Cyltezo(adalimumab-adbm)는 세계에서 가장 가치 있는 약품인 AbbVie의 류마티스 관절염 치료제 휴미라에 대한 바이오시밀러이다. 이 약물들은 둘 다 상호 교환 가능으로 지정되었다. 즉, 오리지널 의약품 처방이 처방자의 개입 없이 자동으로 더 저렴한 바이오시밀러로 채워질 수 있는 것이다(일부 주법에서는 처방자에게 약물 전환에 대해 알려야 함).
이러한 지정이 중요하다. 즉 유럽에 비해 미국의 바이오시밀러 도입은 공격적인 혁신 전술(예: 배제 계약)과 자동 대체의 부재를 이용하는 복잡한 리베이트 규칙으로 인해 방해를 받았다. 호환성은 이러한 장벽 중 일부를 제거하고 가격을 낮추는 데 도움이 될 것이다. 저분자 제네릭에서 볼 수 있는 80-90%가 아니라 30-40%로, 이는 생물학적 제제의 높은 가격을 감안할 때 의약품 지출을 줄이기에 충분하다. (Humira는 출시 후 거의 20년이 지난 2020년에 160억 달러의 수익을 올렸다). Sanford Bernstein 분석가인 로니 갈(Ronny Gal)은 Cyltezo의 이러한 지명을 이 분야의 “이정표적인 성과”라고 불렀다. AbbVie의 변호사는 2023년까지 Cyltezo와 6개의 다른 Humira 바이오시밀러의 FDA 승인을 막았다. 그러나 유럽에서 Humira의 바이오시밀러 버전은 2018년 출시 후 1년도 채 되지 않아 환자 3분의 1 이상을 치료하는 약물이 되었다.
삼성바이오에피스와 바이오젠의 세 번째 바이오시밀러 Byooviz (ranibizumab-nuna)는 로슈의 VEGF(혈관내피세포성장인자) 표적 습성 노화 관련 황반변성 약물인 Lucentis를 참조하여 2021년 미국과 유럽 연합의 안과 분야에서 최초로 승인된 바이오시밀러 바이오의약품이 되었다.
이러한 바이오시밀러는 100번째 오리지날 단일클론항체 승인이 난 같은 해에 처음 출시되었다. 이식된 신장이 거부되는 것을 방지하기 위한 Janssen의 muromonab-CD3(Orthoclone OKT3)의 승인이 거부된 3년 반 후, 불일치 복구-결핍(mismatch repair-deficient (dMMR) 재발성 또는 진행성 자궁내막암 (advanced endometrial cancer)에 대해 GlaxoSmithKline의 programmed cell death receptor-1 (PD-1) 차단제 Jemperli(dostarlimab-gxly)가 승인되었다. 그해 말에 이 약물은 다른 진행성 고형 종양으로 확대되었다. 미국에서 고형 종양 암의 약 14%가 dMMR 돌연변이를 갖고 있는 것으로 알려져 있다.
2021년에 8개의 새로운 FDA 승인 적응증을 받은 Merck의 Keytruda(펨브롤리주맙)가 여전히 지배하고 있는 경쟁이 치열한 분야에서 Jemperli는 일곱 번째 PD-1 차단제 항체이다. OCE의 Richard Pazdur 국장도 이 분야에서 과밀화에 대해 경고하고 있다. 33개 이상의 서로 다른 PD-1 또는 PD-L1 항체에 대한 2,000건 이상의 임상 시험이 있으며, 조정이 거의 없고 비교 연구도 훨씬 적은 실정이다. 12월 15일자 New England Journal of Medicine 에서 그는 “The Wild West of checkpoint inhibitor development.” 라는 제목의 기사에서 이러한 과열된 양상을 지적했다.
Jemperli는 2020년 후반에 COVID-19 관련 현장 검사 지연으로 타격을 받았지만 이 약물은 여전히 암 치료법의 전형적인 빠른 승인 절차의 혜택을 받았다. 그것은 획기적인 지정(가용한 약물에 비해 임상적으로 유의한 평가변수에서 상당한 개선을 보여주는 예비 임상 증거)(그림 2 및 3)을 가졌고 가속 승인을 받았으며 자궁내막암의 경우 RTOR(Real-Time Oncology Review) 파일럿 프로그램을 사용했다. 이는 조기 데이터 제출을 가능하게 하여 평균 검토 시간을 3개월 단축할 수 있었다.
항체 약물의 확장
Jemperli와 같은 항체 약물은 2021년 모든 FDA 승인의 5분의 1을 차지했다. 생물학적 제제는 전체 승인의 40% 미만을 차지했다. 혁신가들은 편리한 항체 제형 및 전달 메커니즘을 통해 가치 있는 항체 프랜차이즈를 계속 발전시키고 있다. 로슈의 1년 2회 안구 임플란트 Susvimo (ranibizumab)는 대조약 Lucentis와 마찬가지로 월 1회 투여가 필요한 삼성과 바이오젠의 Byooviz 이후 1개월 만에 습성 연령 관련 황반변성 치료제로 승인됐다.
매우 높은 LDL 콜레스테롤을 특징으로 하는 희귀 유전 질환인 동형접합 가족성 고콜레스테롤혈증 (homozygous familial hypercholesterolemia)에 대해 Regeneron의 Evkeeza(evinacumab-dgnb)가 승인되면서 심혈관 질환으로의 항체의 느린 확장은 2021년에도 계속되었다. Evkeeza는 지질 대사에 작용하는 angiopoietin-like 3 (ANGPTL3)에 결합하고 이를 차단하는 최초의 항체이다. Regeneron의 VelocImmune 엔지니어링 마우스 플랫폼을 사용하여 개발되었으며, 심혈관 질환에 초점을 둔 다른 두 가지 항체들 중 하나인 PCSK9 억제제인 Praluent(alirocumab)가 있다. 암젠의 Repatha (evolocumab) 가 세번째 항체 약물이다. Evkeeza는 또한 급성 췌장염 예방을 위한 2상을 진행 중이다.
한편, antibody–drug conjugates (ADC)는 2020년에 여러 건, 2021년에 2건의 승인과 함께 불안정한 역사를 거쳐 성숙해졌다. ADC Therapeutics의 Zynlonta(loncastuximab tesirine)는 지난 4월 10번째 ADC가 되었으며, 미만성 거대 B 세포 림프종 (diffuse large B cell lymphoma)을 치료하기 위해 pyrrolobenzodiazepine의 강력한 세포독성 페이로드로 세포 표면 항원 CD19를 처음으로 표적화했다. 절단 가능한 링커는 valine–alanine 과 maleimide이다.
Seagen과 Genmab의 ADC Tivdak(tisotumab vedotin)은 최초의 조직 인자(Tissue factor, TF) 표적 치료제이다. 재발성 또는 전이성 자궁경부암에 대해 승인되었다. 단일클론항체는 미세소관 억제제인 monomethyl auristatin E 와 연결돼 표면 TF 단백질을 통해 암세포에 독성 약물을 전달한다.
CAR-T 세포 치료제
CD19가 아닌 다른 표적을 겨냥한 최초의 키메라 항원 수용체(CAR)-T 세포 치료제가 2021년에 나왔다. Bristol Myers Squibb와 Bluebird Bio의 Abecma(idecabtagene vicleucel)는 대부분의 다발성 골수종 세포에서 발현되는 단백질인 B 세포 성숙 항원(B cell maturation antigen, BCMA)을 공격한다. 2020년 데이터 부족으로 타격을 받은 후, Abecma는 127명의 환자 단일군 연구를 기반으로 최소 4회 요법 으로 재발성 또는 불응성 다발성 골수종에 대해 승인을 받았다. 이 치료법은 지난 8월 유럽연합(EU)에서 조건부 승인을 받았다.
Bristol Myers Squibb의 CD-19을 타깃하는 Breyanzi(lisocabtagene maraleucel)이 지난 5월 미만성 거대 B 세포 림프종에 대한 미국 승인을 받아서 현재 미국에서는 승인된 5가지 CAR-T 세포 치료제가 있다. (Bryanzi는 유럽에서 검토 중입니다). 2022년 2월 FDA는 Janssen과 Legend Biotech의 BCMA를 표적으로 하는 cilta-cel로 알려진 ciltacabtagene autoleucel에 대해 결정이 내려지면 이 분야 세포 치료제는 6개로 증가할 것으로 전망된다. 초기 데이터에 따르면 Abecma보다 훨씬 더 효과적일 수 있으며, 97명의 환자를 대상으로 한 연구에서 이들 중 2/3가 18개월 후에도 질병 진행이 없는 것으로 나타났다.
CAR-T 요법은 대체 요법에 실패한 B 세포 림프종 및 백혈병 환자에게 공격적인 화학 요법을 피하면서 잠재적으로 오래 지속되는 이점을 가진 치료 옵션을 제공한다. CAR-T 치료제는 여러 신속 승인에 적합하며 복잡하고 고도로 개인화된 제조 및 관리로 인해 고비용의 치료제이다.
현재 CAR-T 세포 요법은 수백 명의 환자에게만 도움이 된다. 제조하는 시간을 단축하고 내약성과 편의성을 개선하기 위해 일부 개발자는 동종(기증자 유래) CAR-T 및 CAR-NK(자연 살해) 세포와 같은 차세대 치료제를 개발하고 있다. 비용은 이 치료제의 확장이 직면한 또 다른 장애물이다. 스탓업 회사인 Cellares는 기존의 세포 치료제를 보다 저렴하게 만들 수 있도록 설계된 확장 가능한 제조 플랫폼을 개발하는 회사 중 하나이다.
세포 치료를 위한 CBER 강화
조직 기반 제품 및 백신과 함께 세포 및 유전자 요법은 FDA의 생물제제 평가 및 연구 센터(Center for Biologics Evaluation and Research, CBER)에서 검토된다. McCaughan은 CBER의 직원들이 복잡한 세포 및 유전자 치료 응용 프로그램으로 "COVID-19 이전에 이미 스트레스를 받았다"고 말했다. 팬데믹 백신 승인은 검토자들에게 더 많은 요구를 했다. FDA의 백신 연구 및 검토실(Office of Vaccines Research & Review) 국장 및 부국장은 모두 정부가 발표한 백신 부스터 출시 일정과 FDA 직원이 실현 가능하다고 생각한 일정 사이의 견해 차이로 인해 2021년 하반기에 사임했다.
유전자 및 세포 치료 후원자들은 한 해 동안 커뮤니케이션과 조언의 질이 떨어졌다고 보고했으며, Breyanzi와 Jemperli에 대한 제조 현장 검사도 연기되었다. CBER의 책임자인 Peter Marks는 "우린 우선 순위를 정해야 했고 분명히 COVID-19가 목록의 1위였습니다." "비공식 후원자 회의가 어려움을 겪었습니다." 라고 말했다.
더 많은 세포 및 유전자 치료 신청을 준비하기 위해 CBER을 확장하는 것은 2023-2027년 FDA의 처방약 사용자 수수료법(Prescription Drug User Fee Act, PDUFA VII) 재승인 목표 및 약속 내에서 분명한 목표이다. 10년에 두 번 발표되는 PDUFA 목표는 FDA가 연방 및 산업 기금을 받는 데 필요하다. 새로운 CBER 직원은 132명으로 이 수치는 약물 평가 및 연구 센터가 추가할 인원의 거의 두 배이다. Marks는 이미 "가능한 한 많은 인력을 충원하려고 노력하고 있습니다."라고 말했다. 또한 원격 근무를 통해 지원자 풀을 넓히고 있다. 이러한 조치는 환자를 위해 변화를 만들고자 하는 지친 최전선 임상의를 위한 대체 방안이 될 것으로 전망된다.
현재 대규모 지연 현상이 있는 제조 현장 검사도 디지털 작업 패턴을 보다 광범위하고 영구적으로 수용함으로써 혜택을 볼 것이다. 일반적으로 신약 신청의 약 5분의 1은 사전 승인 현장 조사가 필요하다. 의약품 제조 평가 사무국(Office of Pharmaceutical Manufacturing Assessment) 국장 Stelios Tsinontides에 따르면 원격 검사에 의존하는 외국 파트너 및 제조업체의 규정 준수 기록은 전염병 기간 동안 사전 승인 검사를 55% 단축했다고 전했다.
직원을 늘리는 것과 함께 FDA는 비즈니스 및 정보 시스템을 현대화 및 디지털화하고 서류 작업을 줄임으로써 훨씬 더 효율적이기를 바라고 있다.
너무 빠른 신약 승인
FDA의 2023-2027 목표에는 승인을 가속화하기 위한 추가 노력이 포함된다. 전체 결과가 제공되기 전에 데이터를 순차적으로 제출할 수 있는 3년 된 RTOR 프로그램은 STAR(Split Real Time Application Review) 파일럿을 통해 종양학을 넘어 확장될 것으로 전망된다. 희귀 질환의 최종 목표치에 대한 더 많은 논의가 있을 것이며 잠재적으로 이에 대한 유연성도 있을 것이다.
그러나 FDA는 여전히 대부분 종양학에 사용되는 가속화된 승인 경로를 정리해야 한다. Aduhelm은 불행한 예외였다. 특히 2021년에 필요한 확인 재판을 추적하지 못해 큰 비난을 받았다. 2021년 8월 영국 의학 저널(British Medical Journal)에 발표된 가속화된 암 약물 승인에 대한 후향적 연구에서 지적한 바와 같이 많은 경우 발생하지 않으며 발생하더라도 부정적인 결과가 승인 철회를 촉발하지 않을 수 있다.
FDA는 2021년에 다양한 절차 과정을 정리하기 시작했다. OCE 책임자 Richard Pazdur의 프로젝트 확인은 계속해서 검증되고 철회된 약물을 포함하여 가속화된 승인 하에 승인된 적응증에 관한 정보를 공개적으로 이용 가능하고 검색 가능한 데이터베이스로 제공하고 있다. 무엇보다도 공개적으로 알려진 것처럼 다양한 노력을 통해 2021년에 기록적인 9개의 ‘승인 결정 유보’ 표시가 철회되었다. 여기에는 Secura Bio사의 histone deacetylase inhibitor, Farydak (panobinostat)이 3차 다발성 골수종에, OncoPeptides의 Pepaxto (melphalan flufenamide)가 5차 다발성 골수종에, 3차 위암 적응증인 키트루다(Keytruda)가 있다. Pazdur는 2021년 12월 청문회에서 상원 의원들에게 “나는 가속화된 승인의 팬입니다. 그러나 "시장에서 사용되는 이러한 제품을 평가하려면 더 나은 시스템이 필요합니다."라고 증언했다.
다른 곳의 규제 기관은 희귀 질환들이나 적절한 치료제가 제공되지 않는 질환들을 치료하는 약물에 대해 신속하게 사용할 수 있도록 과정을 가속화하고 있다. 2021년 1월, 영국 의약품 및 의료 제품 규제 기관(Medicines and Healthcare Products Regulatory Agency, MHRA)은 영국이 유럽 연합에서 탈퇴한 후 개발자에게 어필하기 위해 ILAP(Innovative Licensing and Access Pathway)를 시작했다. ILAP는 이런 과정을 가속화하기 위해 지속적으로 데이터를 받아 드리고 있다. 첫 번째 신청은 Merck 의 Welireg(belzutifan)로 von Hippel–Lindau과 관련된 암으로서 다발성 장기에서 (보통 양성) 종양 성장을 특징으로 하는 희귀 질환이다. 이 약은 동일한 적응증으로 2021년 FDA 승인도 받았다. 2021년 1분기에 다른 9개가 그 뒤를 이었다. 수수료는 가입 후보자의 개발 단계에 따라 약 $10,000이 소요된다.
효과적인 의약품에 대한 환자의 더 빠른 접근에 대해 이의를 제기하는 사람은 거의 없다. 코로나바이러스 대유행은 학계, 업계 및 규제 기관이 공중 보건 위기를 해결하기 위해 얼마나 신속하고 효과적으로 동원할 수 있는지 보여주었다. 바이러스성 호흡기 질환이 계속해서 삶과 경제를 황폐화하고 있지만 일부 사람들은 비만이나 기타 전염병과 같이 덜 알려져 있지만 더 널리 퍼진 상태의 질병들에 대한 약물을 개발하도록 인센티브를 제공해야 할 때라고 생각하고 있다.
희귀의약품 지정 및 기타 프레임워크는 환자 수가 적은 희귀 질환에 대한 약물 허가의 속도를 가속화했다. 다음 이슈는 주요 공중 보건 위험에 대해 동등한 조치를 취하는 것이다. 내분비학자 및 비만 약물 개발자들은 CMS가 비만 약물에 대한 약값의 상환을 원한다. 구독 지불 모델 (subscription payment model)을 통해 새로운 항균제의 개발을 촉진하기 위해 미국 파스퇴르법 (US PASTEUR Act)을 통과시키는 것은 그 방향으로 가는 또 다른 단계가 될 것이다. 이 법안은 2021년 6월에 다시 제출되었지만 Capital Alpha의 Smith에 따르면 이번 의회나 다음 의회에서도 진전이 없을 것 같다. 따라서 구독 결제 모델이 미칠 수 있는 영향에 대한 불확실성이 예상되고 실정이다.
사회적으로 가장 시급한 만성 신경퇴행성 질환 중 하나를 해결하기 위해 거의 20년 만에 처음으로 신약인 Aduhelm을 승인한 2021년의 승인은 역효과를 냈다. Prevision Policy의 McCaughan은 "Aduhelm은 종양학 분야의 사람들처럼 생각 하려는 신경 관련 커뮤니티의 노력에 따라 승인된 두 가지 비암 가속 약물들 중 하나였다” 라고 평가했다. 최근 CMS는 임상 시험에 참여하는 소수의 환자만을 대상으로 하라고 제안했고 많은 증거를 요구하며 규제 기관과 같은 역할을 하고 있다. 최종 적용범위 결정은 4월로 예정돼 있지만 논란이 수그러들지 않을 전망이다.